Scientific Report
Period: January - December 2021
O1 - |
The photochemistry of dopamine and its oligomers; |
M01 - |
Mapping the photochemical behavior of DA using combined experimental and theoretical investigations; "D(3)" |
M02 - |
Task 1: Mapping the photochemical behavior of PDA oligomers with different polymeric units using combined experimental and theoretical investigations; "D(10)" |
M03 - |
Drawing general conclusions about the photochemical behavior of DA and PDA oligomers with different polymeric units, as well as project dissemination (Conference participation, progress report, project web page development); "D(12)" |
I. Introduction
The study of the photochemical behavior of a molecule by ab initio type theoretical methods involves calculations for: i) finding of equilibrium geometries for both the fundamental electronic state and the excited electronic states; ii) determining the normal vibration modes for both the basic electronic state and the excited electronic states; iii) the location on the hypersurface of the potential energy of the intersection points (known in the scientific literature as "conical intersection" or CI); determination of absorption and fluorescence spectra which also include vibronic effects on the transitions between the electronic states involved. Choosing an appropriate method is very important to obtain theoretical results that can later be compared with experimental values and through this validation can be used to provide scientific explanations for the phenomena studied. Density Functional Theory (DFT) offers many technical solutions for determining equilibrium geometries, and the time-dependent variant (or TDDFT) for determining the energies of excited electronic states.
II. Working Methodology
Using ωB97X-D3 which is a functional DFT of the range-separated hybrid GGA type whose exchange component consists of a mixture between the Hartree-Fock exchange interaction and that of DFT and SCS-PBE-QIDH belonging to the class of the "double-hybrid" type functional, where in addition to the mixed form of the replacement component and the correlation one, there is also a mixed one between the DFT type correlation and the one derived from the second order theory of Möller-Plesset (or MP2) type we initiated various theoretical calculations to determine the vertical energies of the electronic excitation, the equilibrium geometry for the excited states, respectively the location of the conical intersection points between the fundamental state (S0) and the first excited electronic state (S1). The geometric configuration of the benzene-uracil (or BU) binary structure is shown in Figure 1.
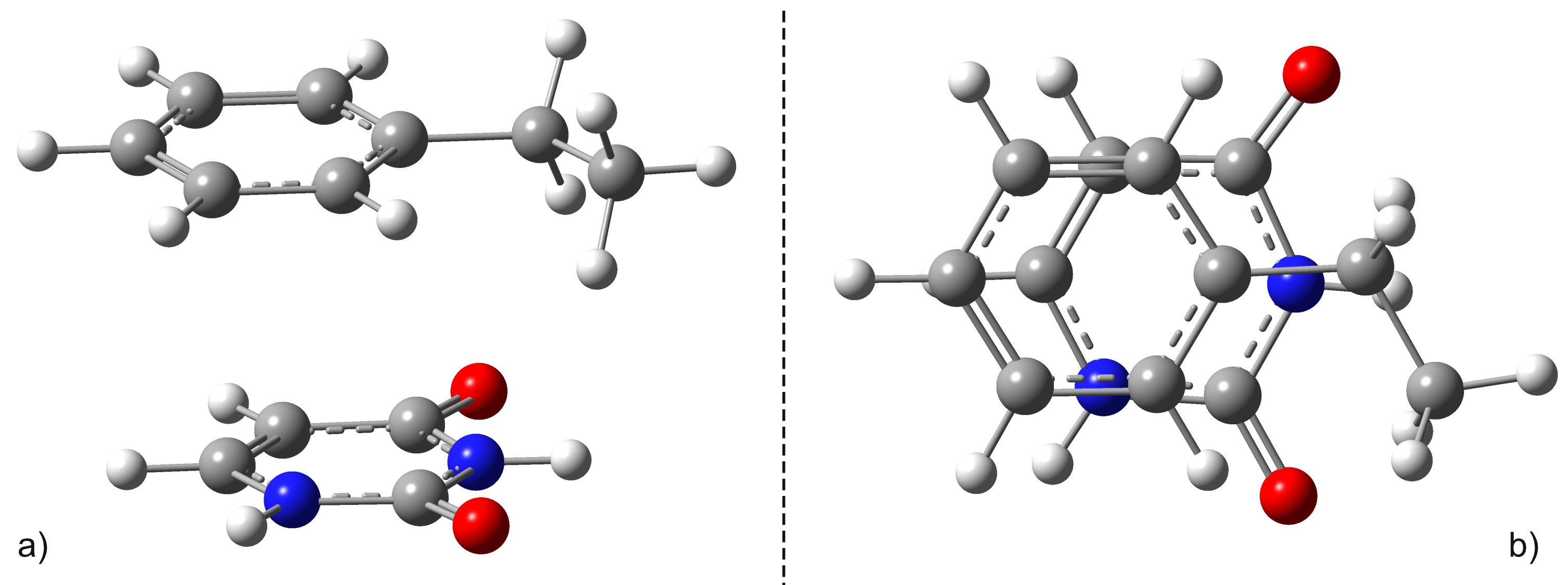
Figure 1. Side a) and top b) views of the "sandwich" (or stacking) type configuration of the uracil-benzene dimer.
As can be seen in Figure 1, the geometric configuration of the binary complex has a sandwich or stacking shape as is better known in the literature. It is important to note that the sandwich configuration of the two aromatic rings is not perfectly plane-parallel, the shortest interplanar distance is 3.435 Å, while the largest is 3.640 Å. The intermolecular interaction energy between the benzene and uracil units is -10.75 kcal/mol obtained considering the same level of theory used to optimize the geometry. The energy value obtained at the DFT level fits very well with the energy of -11.17 kcal/mol calculated taking into account the DLPNO-CCSD (T) theory. The first three low-lying electron excited states for the B − U sandwich complex calculated at the TD-ωB97X-D3/ma-def2-TZVPP theory are: S1 = 233 nm, S2 = 226 nm, and S3 = 218 nm, respectively. Since, in the case of the functional XC of the ωB97X type, the dispersion effects are not explicitly taken into account in the correlation part of the functional, it is only an empirical post-correction of the energy calculation, the same three first low-lying electronic excited states calculated at this time using the TD-SCS-PBE-QIDH/ma-def2-TZVPP theory level are: S1 = 241 nm, S2 = 226 nm, and S3 = 214 nm, respectively. It is well known that due to the wrong dimensionality of the branch space, the standard TDDFT fails near a S0⊗S1 and fails to correctly calculate the geometries of the conical intersection. On the other hand, it has already been shown that the dynamic correlation of electrons counted by scattering effects is essential in the correct description of the supramolecular complex. Therefore, the simple application of multi-reference HF methods without any correction of the type of perturbation or configuration interaction will also lead to an incorrect description. In this regard, the spin-flip (SF) approach of TDDFT provides a more balanced description of the branch space topology. Consequently, the geometry of S0⊗S1 was re-optimized taking into account the SF-TDDFT method. The major difference can be seen in the orientation of the uracil fragment compared to the benzene unit. Specifically, the new link between uracil and benzene units changes from 1.659 Å obtained for the original case to 1.591 Å for the SF case, and rotates the uracil fragment by 20 degrees in addition to the benzene ring. In addition, from an energy point of view, SP-TDDFT also reduces the height of the energy barrier Δ(Estack-ECI) by ≈20 kcal/mol. Despite the differences, standard TDDFT can also estimate an almost good S0⊗S1 geometry, but of course a more precise method, such as spin-flip TDDFT, is needed to accurately describe them.
III. Experimental investigation of the photochemical behavior of dopamine and dopamine-o-quinone
The emission and excitation spectra characteristic of a concentrated aqueous solution of dopamine (10 mg dopamine in 10 ml H2O) have a maximum at 317 nm, and the excitation spectrum has two maximums at 264 and 290 nm. Next, we aimed to investigate the polymerization of dopamine in the presence of a small amount of NaOH and the irradiation of the sample at 264 nm. For this, 150 μl of aqueous dopamine solution was collected over which 45 μl of 0.05M NaOH was added and the emission spectra were recorded under the same conditions immediately after the addition of the NaOH solution. The first emission spectrum acquired after completion with NaOH and irradiation of the sample for 60s shows a drastic decrease in the emission band characteristic of dopamine from 317 nm, accompanied by a redshift of 26 nm. After irradiation at 264 nm for another 60 s, the intensity of this band continues to decrease. At the same time, in the visible region of the emission spectrum, two additional bands appear at 416 and 467 nm. The intensity of the emission of these bands increases with the irradiation and induction of dopamine polymerization. The last spectrum was acquired after a period of about a few minutes. It is observed that the emission band intensity decreases from 343 nm and that the emission bands in the visible region of the spectrum continue to increase. The samples were further investigated using time-resolved fluorescence spectroscopy. In short, the time correlated single photon counting (TCSPC) method involves exciting the sample with a short laser pulse and emitting a fluorescence photon after a period of time dt. For these measurements, a Yb: KGW (Pharos, Light Conversion) femtosecond laser system was used that emits pulses with a duration of 170 fs at 1030 nm. The laser repetition rate is 80 kHz and the pulse energy is 75 μJ. The 1030 nm pulse is used to pump a collinear parametric optical amplifier (Orpheus, Light Conversion), which produces an adjustable output between 620-2600 nm. Additional frequency mixing units can generate the second and fourth harmonics of the signal and free beams, extending the total emission range up to 210 nm. The 260 nm pulses used to excite the dopamine solutions were generated in the additional frequency mixing units by doubling in frequency the 520 nm secondary harmonic of the initial 1040 nm beam obtained in Orpheus. The TCSPC 3-D data matrix represents the intensity of the emitted photons as a function of the emission wavelength and the detection time of the emitted photons relative to the excitation pulse. The results can be analyzed by cutting the 3-D data mat along the wavelength axis at a certain point in time after the excitation pulse, obtaining an emission spectrum.
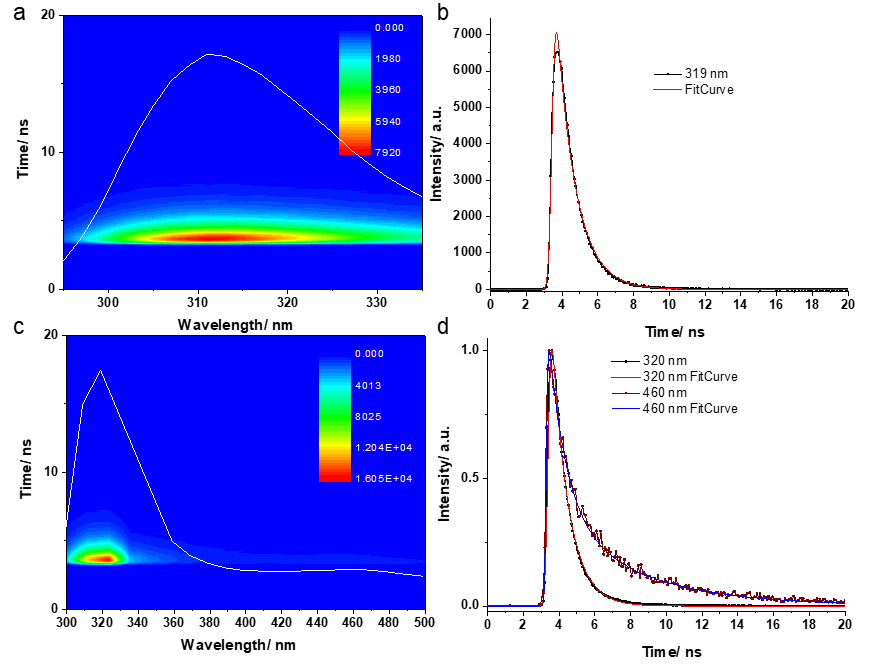
Figure 2. TCSPC 3-d data obtained from the dopamine solution in water (a) and after the addition of a small amount of NaOH (c). The overlapping white lines on the 3-d carpet represent time-integrated fluorescence emission spectra. Normalized kinetic curves recorded at the emission maximums in aqueous dopamine solution (b) and in aqueous dopamine solution after the addition of NaOH (c) together with the fit curves.
The aqueous dopamine sample showed a maximum emission at 319 nm similar to the results of continuous wave fluorescence investigations (See Figure 2). To fit the data we used the GTA method with a single compartment, obtaining a corresponding fit. The lifetime characteristic of the de-excitation of the population of this compartment was calculated at 1.07 ns. After the addition of the NaOH solution, the initial maximum emission showed a redshift of 10 nm and a second maximum emission appeared at longer wavelengths (460 nm), with a much lower intensity. The kinetics of the first maximum emission were similar to those recorded before the addition of the NaOH solution, however, different kinetics were observed for emission wavelengths above 400 nm. For the fitting of the 400-500 nm emission spectral region we used the GTA method, being necessary a scheme with two compartments connected sequentially in order to obtain an adequate fitting of the experimental data. We assumed that the excitatory laser pulse populates a first compartment whose population is then transferred to the second compartment with a time τ1. Next, the population of the second compartment returns to the ground state with a time τ2. The two resulting times had values of 0.98 ns and 4.73 ns.
IV. Theoretical investigation of the photochemical behavior of dopamine and dopamine-q-quinone
Based on the study presented in section II, we demonstrated that in the case of the benzene-uracil test system, the theory of density functionalities, considering an exchange-correlation functional at least of the hybrid-GGA level that contains a mixture between Hartree-Fock-type exchange interaction and the DFT is capable of reproducing both the absorption energy levels and the experimental UV-Vis spectra. At the same time, the theory level TD-ωB97X-D3 / ma-def2-TZVPP is able to reproduce with very good precision the equilibrium geometries for both the fundamental state and the electronic excited states, respectively the frequencies of the normal vibration modes. The only case in which we found a discrepancy is the location of the conical intersection geometries on the hypersurface of the potential energies and for which the spin-flip variant is the one that can give us the correct solution. Taking into account these general conclusions, we performed the optimization of equilibrium geometries, both for the dopamine molecule and for the dopamine-o-quinone variant applying the DFT theory with the exchange-correlation functional ωB97X-D3, respectively the basic set ma-def2-TZVPP. The geometric configurations are shown in Figure 3.
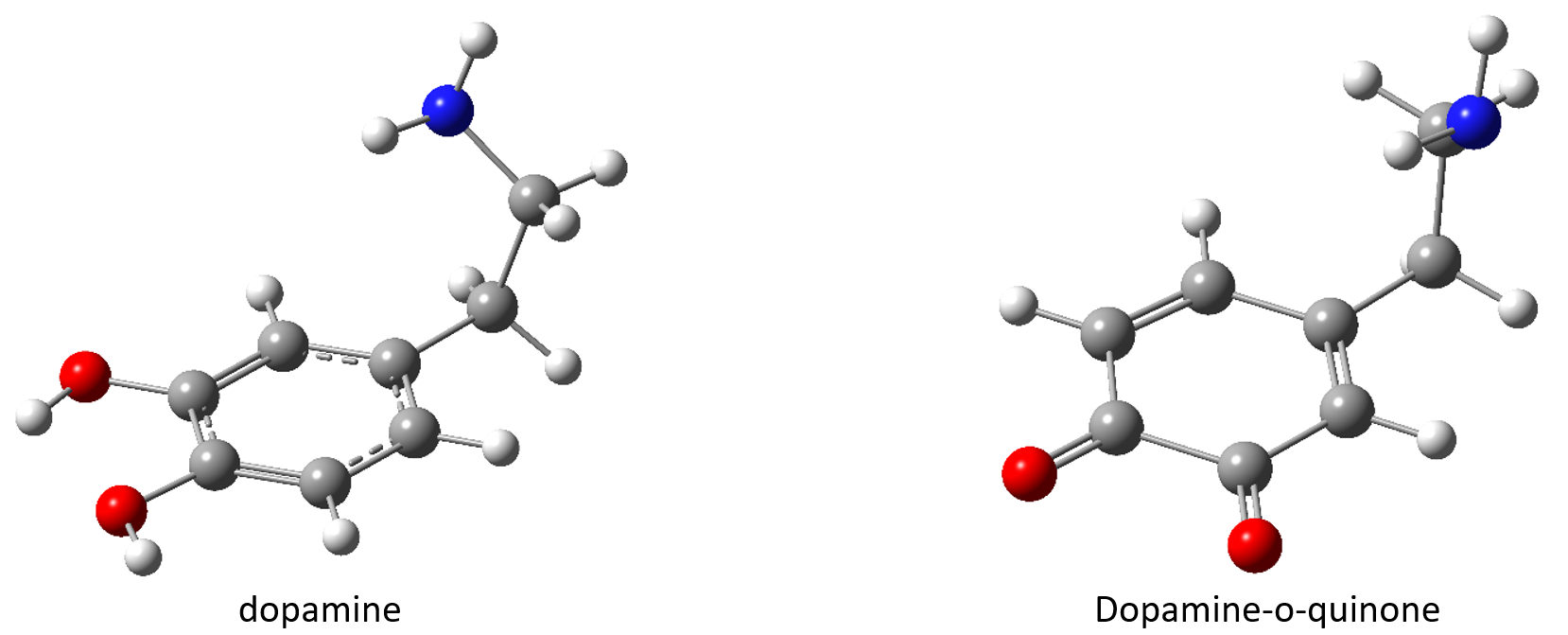
Figure 3. The equilibrium geometries of the fundamental state for the molecular structures dopamine, respectively dopamine-o-quinone obtained at the level of ωB97X-D3/ma-def2-TZVPP theory.
The first three electronically excited states for the dopamine molecule calculated at the TD-ωB97X-D3/ma-def2-TZVPP theory combined with the CPCM (Conductor-like Polarizable Continuum Model) solvent model are: S1 = 240 nm, S2 = 218 nm , respectively S3 = 206 nm, and the absorption peaks measured experimentally are: 290 nm, 264 nm, 237 nm, respectively 220 nm. As can be seen, in terms of absorption peaks, there is a rather significant discrepancy between those measured experimentally and calculated theoretically. As a result, we switched to the use of a more complicated function called SCS-PBE-QIDH, which belongs to the "double-hybrid" type of functional class, and where, in addition to the mixed form of the replacement component and the correlation, there is also a mixed correlation between type DFT and the one from the theory of second order perturbations type MP2. In this case, the first three electronically excited states look like this: S1 = 256 nm, S2 = 224 nm, and S3 = 208 nm, respectively. It is a significant improvement, but still insufficient to have a relatively good match between the values of the frequencies measured experimentally and calculated theoretically. If we move to a higher level of theory defined by the "coupled-cluster" theory and apply the STEOM-DLPNO-CCSD method, we can obtain an even more significant improvement (S1 264 nm, S2 226 nm, respectively S3 204 nm). After determining the absorption spectra, we proceeded to calculate the fluorescence spectra by optimizing the equilibrium geometry of the first excited electronic state. The relaxation of dopamine on the first excited electronic state does not induce major differences in molecule geometry. The fluorescence transition energy considered as the difference of electronic energy taken in the geometric configuration of the equilibrium structures of the first excited electronic state is: 271 nm at the level of the theory TD-ωB97X-D3/ma-def2-TZVPP/CPCM, 285 nm at the level of the theory TD-SCS-PBE-QIDH/ma-def2-TZVPP/CPCM, and for zwitterionic geometric configuration the fluorescence transition energy is: 324 nm at the level of the theory TD-ωB97X-D3/ma-def2-TZVPP/CPCM, 336 nm at the level of TD-SCS-PBE-QIDH/ma-def2-TZVPP/CPCM theory. These values are very close to those measured experimentally, ie 319 nm. Once we obtained the equilibrium geometries of the ground state and the first excited electronic state, respectively we analyzed the normal vibration modes for the two geometries, we proceeded to calculate the fluorescence spectrum with the included vibronic effects, respectively to determine the constant emission rate of fluorescence and implicitly the lifetime of the first excited electronic state. In the case of the dopamine molecule for the constant fluorescence emission rate, the value of 3.17∙108 s-1 was obtained, which means a lifetime of 3.16 ns calculated at the theory level ωB97X-D3/ma-def2-TZVPP/CPCM. If we consider the zwitterionic conformation then for the constant fluorescence emission rate we obtain 1.20∙109 s-1 which gives a lifetime of 0.84 ns being very close to the value 1.07 ns found by experimental techniques. The rate obtained for the case of the neutral molecule is composed in proportion of 81.05% contribution given by the effects of Franck-Condon (or FC) and in proportion of 18.95% given by the effects of Herzberg-Teller (or HT).
Stage summary:
✔
Through the test study we demonstrated the reliability of the DFT method through the functional ωB97X-D3 and SCS-PBE-QIDH in the description of equilibrium geometries for both the fundamental state and the excited electronic states. At the same time, it is not possible to determine the conical intersection points by the time-dependent standard DFT method, it is necessary to apply the spin-flip variant;
✔
UV-Vis and Fluorescence spectroscopy obtained UV absorption and fluorescence spectra for the molecular species dopamine, respectively dopamine-o-quinone. The spectral characteristics specific to the two molecules were interpreted based on theoretical studies using the DFT method;
✔
Using time-resolved fluorescence spectroscopy the fluorescence emission rate constant and the lifetime of the first excited electron state for both the molecular species dopamine and dopamine-o-quinone was obtained which is in very good agreement with the theoretical values found by the DFT methods;
✔
Consideration of the zwitterionic form helps to better assign absorption peaks in the experimental UV-Vis spectrum and to identify the lifetimes of the first excited electronic state;