Scientific Report
Period: January - December 2022
O2 - |
The photochemistry of polydopamine analogues and its higher-level cluster aggregates; |
M04 - |
Task 2: Mapping the photochemical behavior of different oligomer aggregates of PDA using combined experimental and theoretical investigations; "D(17)" |
M05 - |
Task 3: Mapping the photochemical behavior of different oligomer aggregates of PDA analogues using combined experimental and theoretical investigations; "D(22)" |
M06 - |
Drawing general conclusions about the photochemical behavior of different oligomer aggregates of PDA, as well as project dissemination (Conference participation, progress report, scientific article); "D(24)" |
I. Introduction
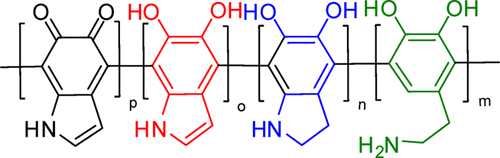
TIn a previous work related to the polymer structure of polydopamine (PDA), it was concluded that the polymer form of polydopamine is not homogeneous, they can have different chemical structures as shown in Figure 1.
Figure 1. Chemical structures of the polymeric units of polydopamine.
The structure of these units has been confirmed based on experimental investigations by IR, Raman and NMR spectroscopy. At the same time, this study also confirmed more complex forms of structural organization but this time between polymer chains and as such different forms of aggregation between polymer chains were proposed. Thus, aggregations were found in the form of stacking (or layered packing) with parallel or antiparallel orientation, H-bond (intermolecular hydrogen bonding), T-shape (or perpendicular) defined by the aromatic rings of dopamine units. All these forms of polymer chain organization, i.e. aggregation of polymer chains will significantly influence the photochemical behavior of PDA compared to those observed in polymer units such as: dopamine (D), dopamine-o-quinone (Q), 1H-indole-5,6-diol (I), 5,6-dihydroxy-indoline (H). It is important to note that the dopamine unit (i.e. D) is the one that is most abundant in the polymer chain because it is not followed by secondary reactions after polymerisation, but the homogeneous form of D-type units cannot be obtained following polymerisation reactions.
II. UV-Vis absorption spectra of PDA
The photochemical behavior of the dopamine unit has already been investigated and characterized in detail in a previous paper . As a result, the absorption behaviour of PDA was investigated. It was measured in different solvent media: TRIS, water (with two different concentrations 1 ml PDA in 1.5 ml water, respectively 0.5 ml PDA in 2 ml water), in PBS and in solvent water mixed with copper sulphate. The absorption spectrum of PDA is shown in Figure 2.
Figure 2. UV-Vis absorption spectra of polidopamine in different solvent media.
As can be seen from Figure 2, the PDA in water solvent in low concentration is the one that differs significantly suggesting that the first step of polymerization predominantly between the aromatic rings of dopamine are followed by secondary reactions. In this next step various proton and charge transfers from hydroxyl groups to amine groups occur, following which other polymeric units with semi-quinone, quinone or protonated amine (NH3+) forms will appear. The presence of absorption even in the 500 - 800 nm spectral range indicates that the electronic excitations are not localized on a single polymeric unit, in general on the dopamine ones, but should have a delocalized form. On the other hand, the fact that in the low dilution form we do not see absorption in the spectral range specified before, tells us that after the first polymerization step we still do not have major modifications of the polymeric units, and that these modifications are generated by different proton and charge transfers, i.e. the closure of the ethylamine fragment and the realization of the indole-like form caused by the subsequent aggregation of the polymer chains. As a result different forms of oligomers (trimers and pentamers) were generated having different shapes of the PDA polymer sequence and they were studied considering DFT methods for equilibrium geometry determination, respectively TDDFT for UV-Vis absorption spectra determination, equilibrium geometry localization in the first excited state, respectively fluorescence frequency determination.
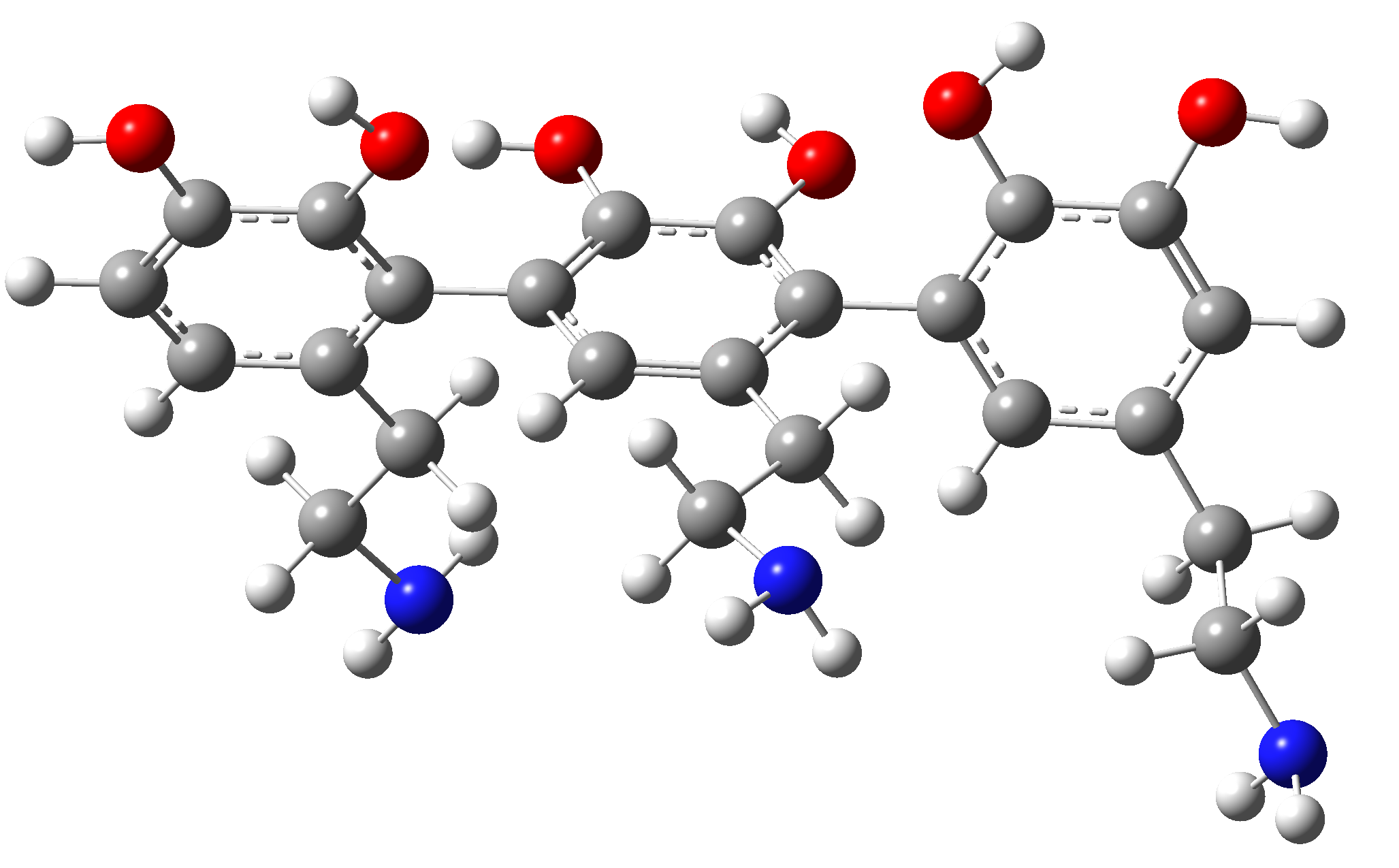
DDD. This trimer contains three dopamine units, it is a homogeneous segment without showing side effects such as proton transfer from hydroxyl to amine. The equilibrium geometry was determined by the DFT method using the exchange-correlation functional ωB97X with empirical correction for dispersion in Grimme's D3 form , using the def2-TZVPP basis set and including the CPCM solvent model where water was considered as the solvent medium. For the determination of the excited electron energies we used the TPSSh exchange-correlation functional with 20% exchange of Hartree-Fock form. The shape of the equilibrium geometry is illustrated in Figure 3. As can be seen, the D-units of the trimer are not co-planar, they are rotated by approx. 110° with respect to each other. After obtaining the equilibrium geometry, the UV-Vis absorption spectrum was determined (see Figure 3).
Figure 3. Equilibrium geometry of the DDD trimer obtained with the ?B97X-D3/def2-TZVPP/CPCM(water) method.
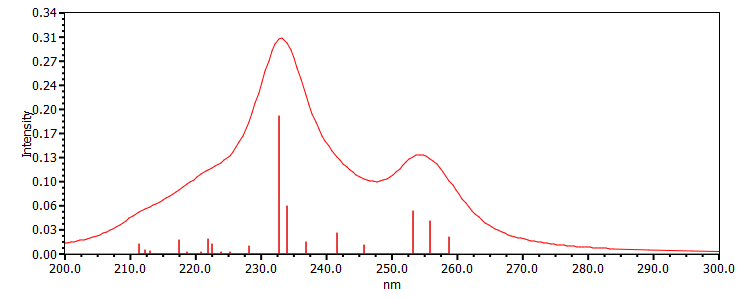
Figure 4. UV-Vis absorption spectrum of the DDD trimer obtained with the TD-TPSSh-D3/def2-TZVPP/CPCM(water) method.
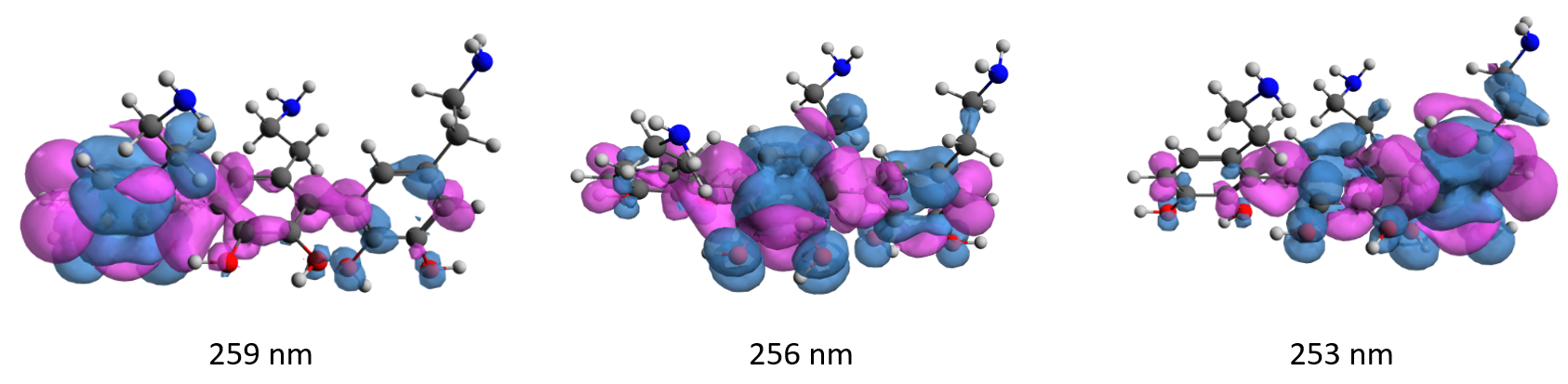
The first spectral peak located in the 250 - 260 nm spectral range is composed of three transitions that have almost the same shape as the S0 S1 transition of dopamine, but they have been slightly shifted due to the interaction of the three D units. These three values are 259, 256 and 253 nm. After obtaining the UV-Vis absorption spectrum, the geometry of the first steady state was also optimized. The only major change is the destruction of the aromatic structure of the benzene ring of the units in the centre of the sequence, i.e. from the value 1.39 Å it changes to 1.39-1.39-1.41-1.39-1.41-1.39 Å. At the same time the fluorescence frequency value changes at 291 nm. If we generate the density differences corresponding to the first three electronic transitions of the DDD trimer (see Figure 5), we observe that the nature of the electronic transitions has a delocalized form, i.e. they represent electron transfer from a lateral D-unit to the central D-unit.
Figure 5. Electron-gol density differences for the three S0 🠢 S1 transition values of the DDD trimer ( blue = void density, pink = excess electron density).
III. Aggregation effects on the nonradiative relaxation of catechol
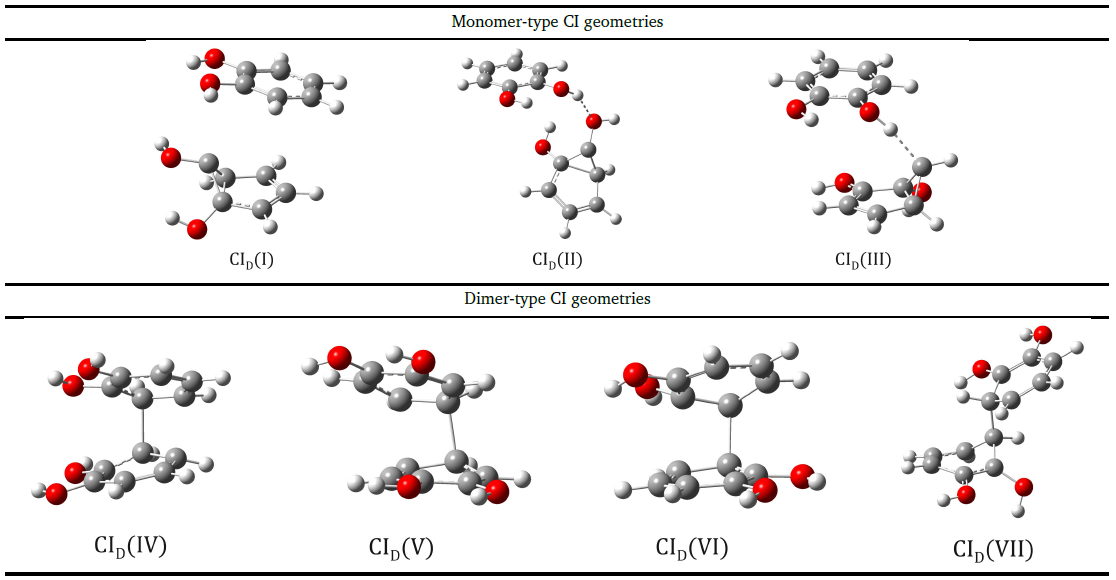
In order to study the aggregation effects of polymer chains on the nonadiabatic relaxation of excited electronic states, the catechol dimer model was chosen, which contains the same aromatic ring and hydroxyl moieties as found in the dopamine molecule. It has already been shown that acetonitrile and cyclohexane solvent media can strongly influence the excited medium lifetime of the excited state even at higher concentrations of catechol. This may be partly related to the different aggregation patterns in the medium of the two solvent media, where cyclohexane is thought to be more favourable to catechol stacking-type aggregation than acetonitrile, which in turn may affect the geometry of the CIs (conical intersection) and the energy levels they generate differently. Accordingly, we investigated how the IC geometries found for monomers change in the presence of aggregation. As a first step, we constructed dimeric structures in which one of the monomers takes the form of the CI geometry. We found three such geometries in which a monomer CI configuration and a planar catechol configuration are combined into a dimeric structure. Of course, given the CIM geometries (I, II or III, where M stands for monomer) and the different relative positions of the planar molecule, more than three dimeric structures we found. The CI geometries of catechol dimers were calculated using SF-ωB97X-D3/madef2-TZVPP/CPCM at the theoretical level (See Figure 6). In the case of the first two CID(I) and CID(II) configurations, it is observed that the CIM(I) structure can be observed, but first in a stacking form, in the second case it is in the form of a hydrogen bond, while in the third CID(III) configuration, the CIM(II) structure is observed forming a special hydrogen bond with a carbon atom. From the analysis of the bonding parameters, it can be concluded that the CIM-type geometry within the dimer retains the semicadrilateral shape already observed for the isolated monomers, while the other monomer retains the non-aromatic ring shape that was characteristic of the ground-state catechol. In terms of energy balance, the CID(I-III) configurations give energy values more than 20 kcal/mol higher than the steady-state geometry in the S1 state equilibrium dimer configuration, calculated in the acetonitrile solvent medium. These energy differences come both from the weaker intermolecular interaction between the monomers and from the strain energy found for the CIM geometries of the isolated monomers compared to the equilibrium geometry in the S1 state.
Figure 6. Structures of CI-type geometries of monomeric and dimeric form obtained with the SF-ωB97X-D3/def2-TZVPP/CPCM(water) method.
At the same time, a similar analysis was made for the case of benzene dimer, where it was found that the non-adiabatic effects of the dimer type depend strongly on the fragments in the equatorial position of the benzene ring, which makes the photochemical behaviour of benzene dimer different from that of catechol (and therefore dopamine). In the case of benzene the conical intersection of CID(VII) shape is the most energetically favourable, while in the case of catechol this geometrical shape is only an "avoided crossing" with an energy difference between the two peaks of 0.007 eV.
Stage summary:

✔
It has been shown by experimental measurements combined with DFT and TD-DFT theoretical studies that the UV-Vis spectra of polydopamine depend on the sequence form of dopamine, dopamine-o-quinone, 1H-indole-5,6-diol, 5,6-dihydroxy-indoline units as well as on the different forms of protonation of amine groups and deprotonation of hydroxyl moieties. The most important influence is the dopamine-o-quinone form which can induce a shift of the energy spectrum from 260 nm to 700 nm;
✔
The energetic and radiative relaxation effects, i.e. the fluorescence effects, are closely related to the central pentamer units or the existence of polymeric units of the dopamine-o-quinone form;
✔
For non-adiabatic relaxation channels, in the case of dimeric catechol, several dimer-like CI geometries have been found in which both monomers exhibit substantial geometrical distortions together with the formation of a weaker C-C bond between the two catechol monomers. These CI geometries are more energetically favorable and, in the case of aggregation processes, the excited states of the catechol are more likely to disintegrate through these non-adiabatic deactivation channels than those found for the monomer;
✔
The non-adiabatic effects of the dimer type, depend strongly on the fragments in the equatorial position of the benzene ring, which makes the photochemical behaviour of the benzene dimer different from that of catechol (and hence dopamine);