Scientific Report
Period: July - December 2017
O1 - |
Validation of the theoretical framework through the comparison with different experimental results; |
M01 - |
Computing the vertical excitation energies and the theoretical UV absorption spectra for the azopyridine functionalized square planar Ni-tetrakis(pentafluorophenyl)-porphyrin (Ni-TPP) complex with five-coordinate metal considering both the "low" and "high" spin configurations as well as searching for the ISC point and computing the spin-orbit couplings; |
M02 - |
Computing the vertical excitation energies and the theoretical UV absorption spectra for the biazopyridine functionalized square planar Ni-tetrakis(pentafluorophenyl)-porphyrin (Ni-TPP) complex with six-coordinate metal considering both the "low" and "high" spin configurations as well as searching for the ISC point and computing the spin-orbit couplings; |
Introduction
Materials with "spin crossover" (SCO) or "spin transition" (ST) properties are of major interest through their potential applications such as molecular memory, sensors or molecular switches1. The basic idea behind this phenomenon is that by an external disturbance, either temperature, pressure or exciting light, the system with a well defined spin state passes into another stable state but with a different spin multiplicity. If the transition is in only one direction these systems fall into the category of materials with ST properties, and if this transition is reversible, they have SCO type properties. The transition from a low spin state (LS) with low multiplicity (eg S = 0) into a higher multiplicity of spins (eg S = 2) called "high spin state "(HS) involves changes in volume, magnetic state or simple the color response of the material2. The novelty lies in the fact that these properties can be exploited to create molecular systems with varying volume manipulated by laser radiation or systems with controllable magnetic states. The energy scheme of the SCO phenomenon is shown in the following figure:
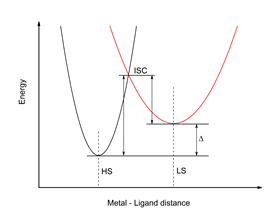
Figure 1. The energy scheme of the SCO phenomenon.
As can be seen from the diagram shown in Figure 1, the transition from a spin to another stable state involves the transition of the potential barrier defined by the isoelectric point of the "intersystem crossing" (ISC). In this case, the external stimulus will only help the system to overcome this barrier. On the other hand, changing HS in LS or vice versa also involves a strong spin coupling so that the transition might be done with good efficiency in both directions. If the potential barrier is relatively high (> 1 eV), the temperature or pressure effects are no longer able to help the system move from a spin state to another. The only possibility to pass this barrier is to use the electronic excited states so that by excitation, the system moves in a near or even higher electronic state to the ISC point from which it can passes much easier into the other spin state (see Figure 2 ). Light Induced Excited Spin State Trapping (LIESST) transitions have been first observed by Decurtins et al3 in 1984 for the Fe-(propyltetrazole)6-(BF4)2 system. Using a radiation source of 514 nm, the system initially in the spin state LS was transferred to another metastable state (T ≈ 1 μs) having the HS state. Two years later, in 1986, one of the authors4 succeeded in inducing the reverse HS → LS transformation using another wavelength of 752.7 nm.
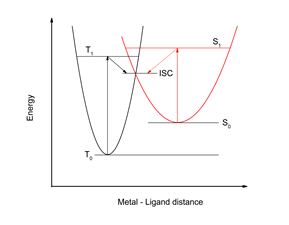
Figure 2. Energy scheme of induced SCO phenomenon by electromagnetic radiation.
Working Methodology
Being supramolecular systems with more than 50-60 atoms, the theoretical methods based on solving the Schrödinger equation and the determination of molecular properties by means of the wave function become almost impossible. Density Functional Theory (DFT)5 can be considered as an alternative solution that describes electronic structure at the electron density (XYZ coordinates) and not the wave function (3N, N = atomic number) level. Within the DFT theory, the accuracy of the description of the molecular systems is given by the form of the exchange-correlation function. Therefore, in order to have a more correct approach, it is necessary to use functionals that include both the effects of the exchange interaction by including the term "exact exchange" and the correlation effects containing both dynamic (correlation between pairs of electrons ) and static (correlation between energetically closed states) correlation terms.
Validation of the Method. To validate the working method, we choose a relatively simple case for which high-cost theoretical methods such as multireference or coupled cluster methods can be successfully applied. From this point of view, the phenothiazine molecule can be considered as a proper choice since it contains aromatic molecular moieties, namely nitrogen or sulfur hetero atoms, on the one hand, and on the other hand is a sufficiently small molecule for which ecuation-of-motion coupled cluster (EOM-CCSD)6, local CC2 (LT-DF-LCC2)7 or multireference self consistent field (MCSCF)8 can be successfully applied. In the case of the phenothiazine molecule, we compared the results obtained by the previously defined three advanced methods with the DFT method considering the functional MN12-SX exchange correlation9. The data for excited electron energies, oscillator strengths (intensity of absorption) for the first three excited states calculated by four different theoretical methods are shown in Table 1.
Table 1. The vertical excitation energy in eV (and nm) and the oscillator strength for the first four excited states of phenothiazine obtained by different theoretical methods.
| Method | S1 | S2 | S3 | S4 |
|---|---|---|---|---|
| MCSCF | 5.39 eV (230 nm) | 5.52 eV (224.7 nm) | 5.71 eV (217.2 nm) | -- |
| 0.0027 | 0.0344 | 0.0607 | ||
| EOM-CCSD | 4.34 eV (285.9 nm) | 4.50 eV (275.5 nm) | 4.64 eV (267.3 nm) | -- |
| 0.0006 | 0.0274 | 0.0497 | ||
| LT-DF-LCC2 | 3.66 eV (338.9 nm) | 3.94 eV (314.7 nm) | 4.10 eV (302.3 nm) | 4.90 eV (252.9 nm) |
| 0.0006 | 0.0271 | 0.0778 | 0.1311 | |
| MN12-SX | 3.80 eV (326.3 nm) | 4.19 eV (296.1 nm) | 4.33 eV (286.3 nm) | 5.21 eV (238.1 nm) |
| 0.0004 | 0.0301 | 0.1090 | 0.1818 | |
| Exp10 | -- | 320 nm | -- | 250 nm |
As it can be seen, the LT-DF-LCC2 method reproduces with good approximation the UV-Vis absorption experimental frequencies. The other advanced methods of MCSCF and EOM-CCSD significantly underestimate the experimental values, so they can not be used to calibrate the DFT method11. The MN12-SX correlation exchange function presents a viable solution for studying excited states by the fact that the energy values of the excited electronics are very close to those obtained by the advanced LT-DF-LCC2 method. The LT-DF-LCC2 energies are slightly underestimated but the difference between the two sets of results is small.
Results
Applications in the case of Ni-Tetrakis(pentafluorophenyl)-porphyrin (Ni-TPP) functionalized with azopiridine (Ni-TPP-AP). In order to obtain even better concordance for the MN12-SX DFT method we have chosen the Ni-TPP-AP molecular complex which shows spin-crossover transition behavior and for which there are experimental data for the UV-Vis absorption spectra in the literature12.
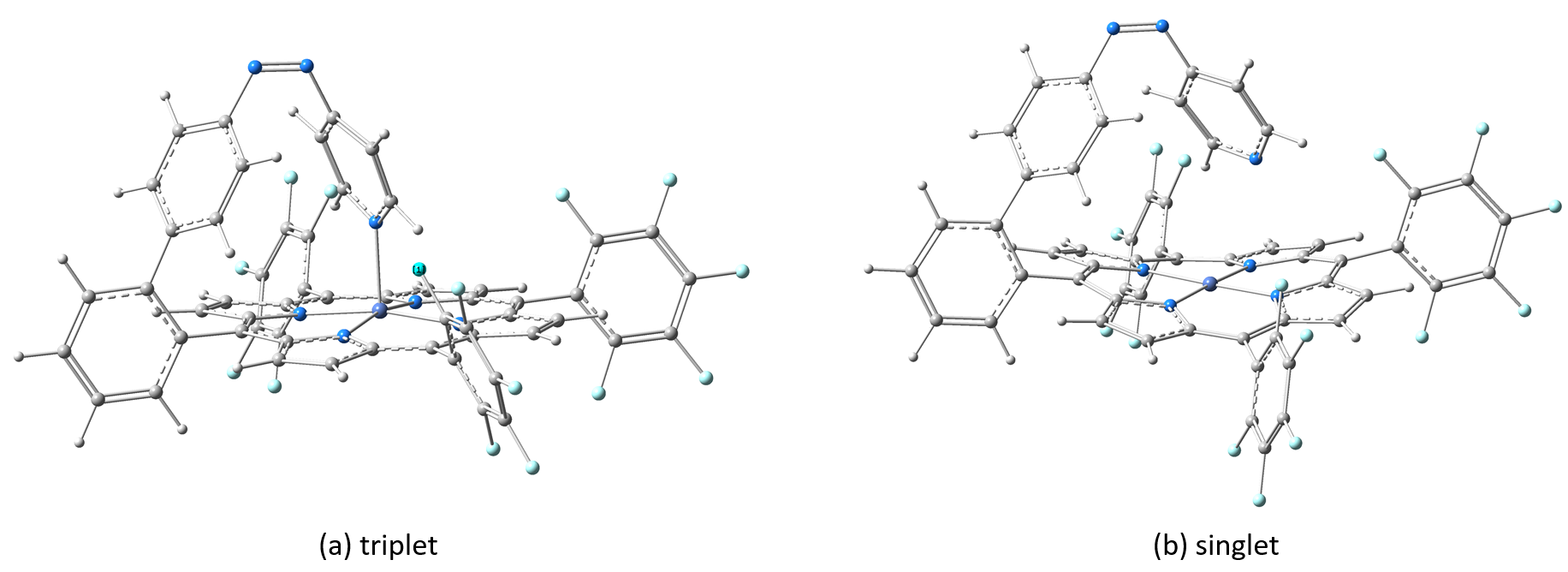
Figure 3. The geometric configuration of the Ni-TPP-AP molecular complex in its two triplets (a) and singlet (b) spin states configuration, respectively.
The equilibrium geometries were optimized in both single and triplet spin configurations and for which we have calculated the theoretical UV-Vis absorption spectra using the MN12-SX/def2-TZVP level of theory and including the first 30 excited electron states. The geometry structures of the Ni-TPP-AP for the two spin states are shown in Figure 3, and their UV-Vis absorption spectra are shown in Figure 4.
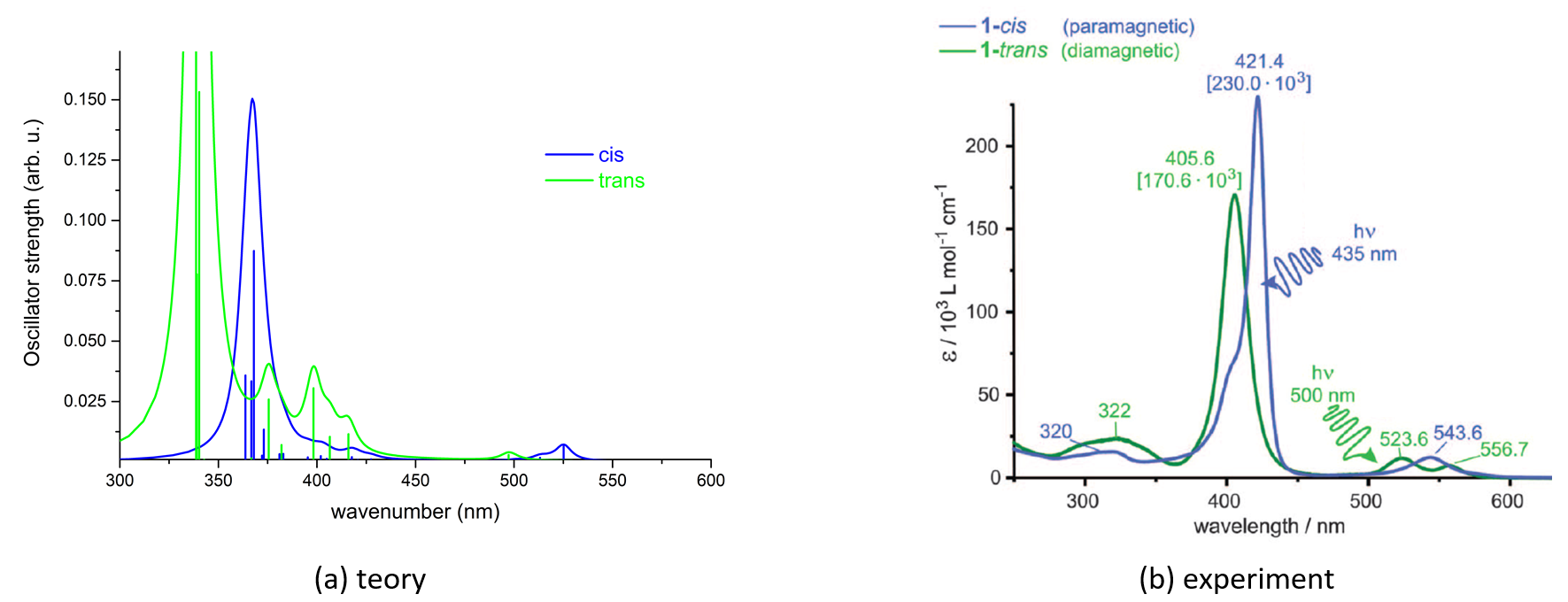
Figure 4. UV absorption spectra for the Ni-TPP-AP molecular complex obtained by theoretical modeling (a) and by experimental techniques10 (b), respectively.
As it can be seen, the theoretical UV-Vis absorption spectrum calculated by the MN12-SX/def2-TZVP level of theory reproduces with a good approximation of the spectral shape and the position of the absorption peaks obtained by experimental techniques. The redshift (the deviation from the experimental values) is on average 30 cm-1. The energy difference (?E = ET - ES) between the two spin states is 0.533 eV in the equilibrium geometry of the triple state, ie -2.017 eV for the equilibrium geometry of the singlet state. The conformational difference between the equilibrium geometries of singlet and triplet spin states is 0.227 eV. In the case of the Ni-TPP-AP molecular complex, we have a pyramidal coordinate configuration, but the octahedral coordination configuration looks also very interesting. For this reason we have constructed another molecular complex, where instead of a single fragment of azopyridine we considered two fragments such that the nitrogen atom of the second azopyridine fragment makes a new coordination bond and at the end we obtain for the nickel atom (II) the octahedral coordination configuration. At the same time, because of the large volume of calculation, we have neglected the three pentafluorophenyl groups (Ni-P-biAP) from the porphyrine ring.
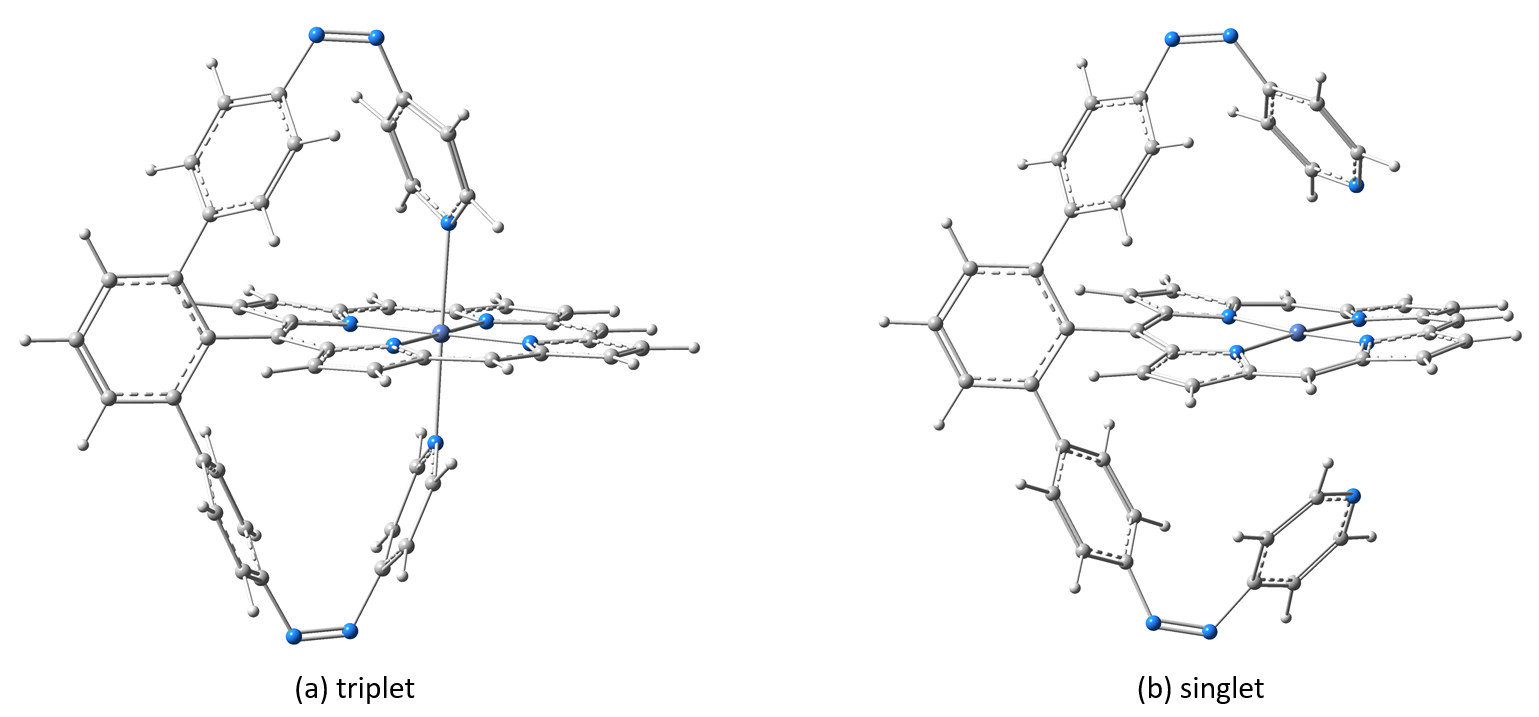
Figure 5. The equilibrium geometry configuration of the Ni-P-biAP molecular complex in the two triplets (a) and singlet (b) spin states, respectively.
Applications for the Ni-P-biAP molecular complex. As in the previous case, we searched for the equilibrium geometries of the Ni-P-biAP molecular complex for both the singlet and triplet spin configurations. After that we generated the theoretical UV-Vis absorption spectra including the first 30 excited electron states. The geometric structures for Ni-TPP-biAP having the two spin states are shown in Figure 5, while the UV-Vis absorption spectra are shown in Figure 6.
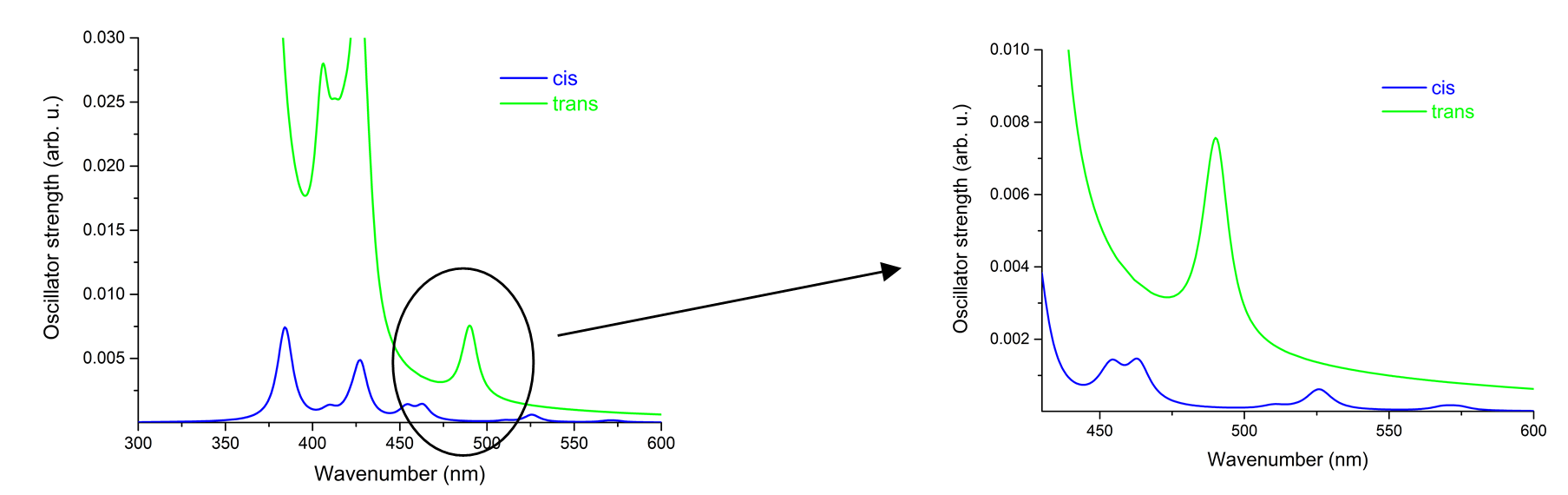
Figure 6. The UV absorption spectrum for the Ni-P-biAP molecular complex obtained by theoretical modeling.
The energy difference (?E = ET - ES) between the two spin states is 0.947 eV for the equilibrium geometry of the triple state, ie -0.978 eV for the equilibrium geometry of the singlet state. The conformational difference between the equilibrium geometries of singlet and triplet spin states is 0.135 eV.
Intersystem crossing points (ISC). Using the algorithm called "Penalty Function"13 we have identified the possible intersection points on the hyper-surface of the potential energies for the two singlet and triplet spin configurations. For the molecular complex Ni-TPP-AP we started the searching procedure for the ISC intersection point where for a given geometry the energies of the singlet and triplet spin configurations are equal. This geometry is characterized by a Ni-N bond lengthen from 2.089 Å of the triplet spin state to 2.257 Å in the case of ISC geometry. The potential barrier specific to this ISC point is 0.273 eV. In the case of the Ni-P-biAP molecular complex, we found the ISC point close to the equilibrium geometry of the triplet spin state geometry. For this intersection point, the Ni-N bonding distance is 2.379 Å compared to the value of 2.276 Å corresponding to the equilibrium geometry of the singlet spin state. The potential barrier specific to this ISC point is 2.063 eV.
Bibliography:
J. A. Real, A. B. Gaspar, M. C. Muñoz, Dalton Trans., 12, 2062 (2005).
A. Hauser, Topics in Current Chemistry, 234, 786 (2004).
S. Decurtins, P. Gutlich, C. P. Kohler, H. Spiering, A. Hauser, Chem. Phys. Lett., 105, 1 (1984)
A. Hauser, Chem. Phys. Lett., 124, 543 (1986).
P. Hohenberg, W. Kohn, Physical Review 136(3B), B864 (1964).
T. Korona and H.-J. Werner, J. Chem. Phys., 118(7), 3006 (2003).
D. Kats, T. Korona and M. Schütz, J. Chem. Phys., 125(10), 104106 (2006).
P. Celani and H.-J. Werner, J. Chem. Phys., 112(13), 5546 (2000).
R. Peveratia and D. G. Truhlar, Phys. Chem. Chem. Phys. 14, 16187 (2012).
J. A. VanAllan, G. A. Reynolds and R. E. Adel, J. Org. Chem. 27, 1659 (1962).
A. Bende, AIP Conference Proceedings, 1917, Article Nr. 020002 (2017).
S. Venkataramani, U. Jana, M. Dommaschk, F. D. Sönnichsen, F. Tuczek, R. Herges, Science, 331, 445 (2011).
C. Ciminelli, G. Granucci and M. Persico, Chem.-Eur. J. 10, 2327 (2004).