Scientific Report
Period: January - December 2018
O1 - |
Validation of the theoretical framework through the comparison with different experimental results; (Continuing from 2017) |
M02 - |
Computing the vertical excitation energies and the theoretical UV absorption spectra for the biazopyridine functionalized square planar Ni-tetrakis(pentafluorophenyl)-porphyrin (Ni-TPP) complex with six-coordinate metal considering both the "low" and "high" spin configurations as well as searching for the ISC point and computing the spin-orbit couplings; (Continuing from 2017) |
M03 - |
Drawing conclusions about the basic mechanism and the role of different molecular excited states in light-driven coordination induced spin state switch as well as project dissemination; |
Preliminary Results
In our preliminary progress report we have presented results about the equilibrium geometry optimization and about the searching of intersystem crossing (ISC) points on the potential energy surfaces of the electronic states with singlet and triplet spin states in the case of Ni-Tetrakis(pentafluorophenyl)-porphyrin functionalized with azopiridine (Ni-TPP-AP) as well as in the case of Ni-porphyrin functionalized with two azopiridines (Ni-P-biAP) molecular complexes. We have also shown that the MN12-SX exchange-correlation DFT functional gives reasonable good results both for the geometrical parameters (bonds, angles) and for the electronic excited state energies with different spin states.
Ground State ISC
The localization of the ISC point is not a simple task. Unfortunately, this searching procedure is not implemented in any standard quantum chemistry program packages. In order to make efficient the searching procedure we have developed a homemade Python code (PyISC) which is able to perform both intersystem crossing and conical intersection point searches either for the ground or the excited state electronic configuration. The logical scheme of our code is shown in Figure 1.
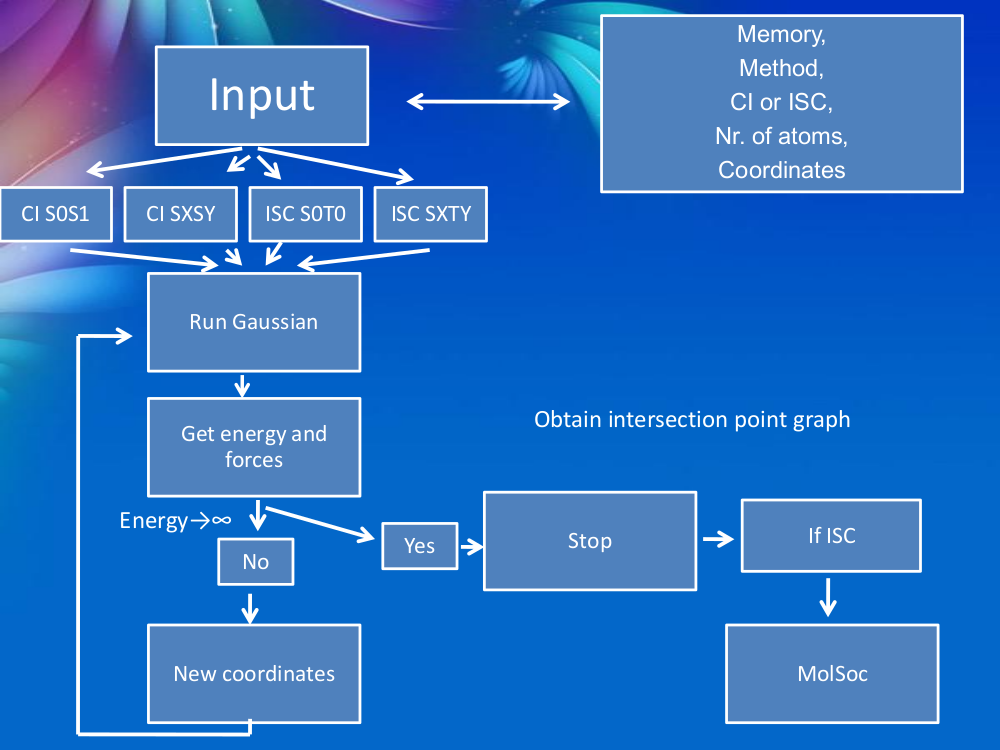
Figure 1. The logical scheme of the PyISC program code.
The mathematical model of the "Penalty Function"1 (PF) was already described in our previous work2. The input parameters of the code are the following: Mem - the amount of the memory (in GB) used by the Gaussian quantum chemistry (QC) program; Method - the QC method and the basis set used by Gaussian; CI or ISCI - the type of the crossing point (CI means "Conical Intersection" or crossing point between ground or excited electronic states having the same spin states and ISC means "InterSystem Crossing" or crossing point between ground or excited electronic states having different spin states); Nr. Atoms - The number of atoms of the molecular system; Coordinates - the Cartesian coordinates of the atoms. Using these input parameters, the code calls the Gaussian QC software for calculating the energies and forces. Then checks the energy gap between the two states and if this gap is still larger than a well defined value, the code generates new coordinates based on the PF method and calls again the Gaussian QC software. If the energy gap is smaller than 0.0001 H the searching procedure stops. In the case of singlet-triplet ground state ISC calculation the spin-orbit coupling is also computed using the MolSOC software3-5.
Ni-TPP-AP. As we already presented in our 2017 yearly report (See Res_2017) an ISC geometry point was located on the hyper-potential energy surface between the triplet and singlet equilibrium geometry configurations. The most relevant parameter which describes this transition state is the d(Ni···N) distance. The variation of this distance shows the following behaviour: for the triplet equilibrium geometry d(Ni···N)=2.089 Å, for the ISC point d(Ni···N)=2.450 Å, while for the singlet equilibrium geometry d(Ni···N)=3.321 Å. The relative energy differences are: 0.227 eV, 0.393 eV and 0.000 eV. One needs to mention that it was found another ISC geometry point characterized by the d(Ni···N)=2.257 Å but its relative energy is 0.524 eV. In order to compare the variation of the d(Ni···N) distance and the position of the ISC points on the potential energy hyper-surface we have performed constrained geometry optimization where we varied step by step the d(Ni···N) distance between the 2.089 - 3.321 Å but keeping this value constant for each geometry optimization. The variation of the relative energy for the triplet and singlet spin states and the position of the ISC points based on different d(Ni···N) distance is presented in Figure 2.
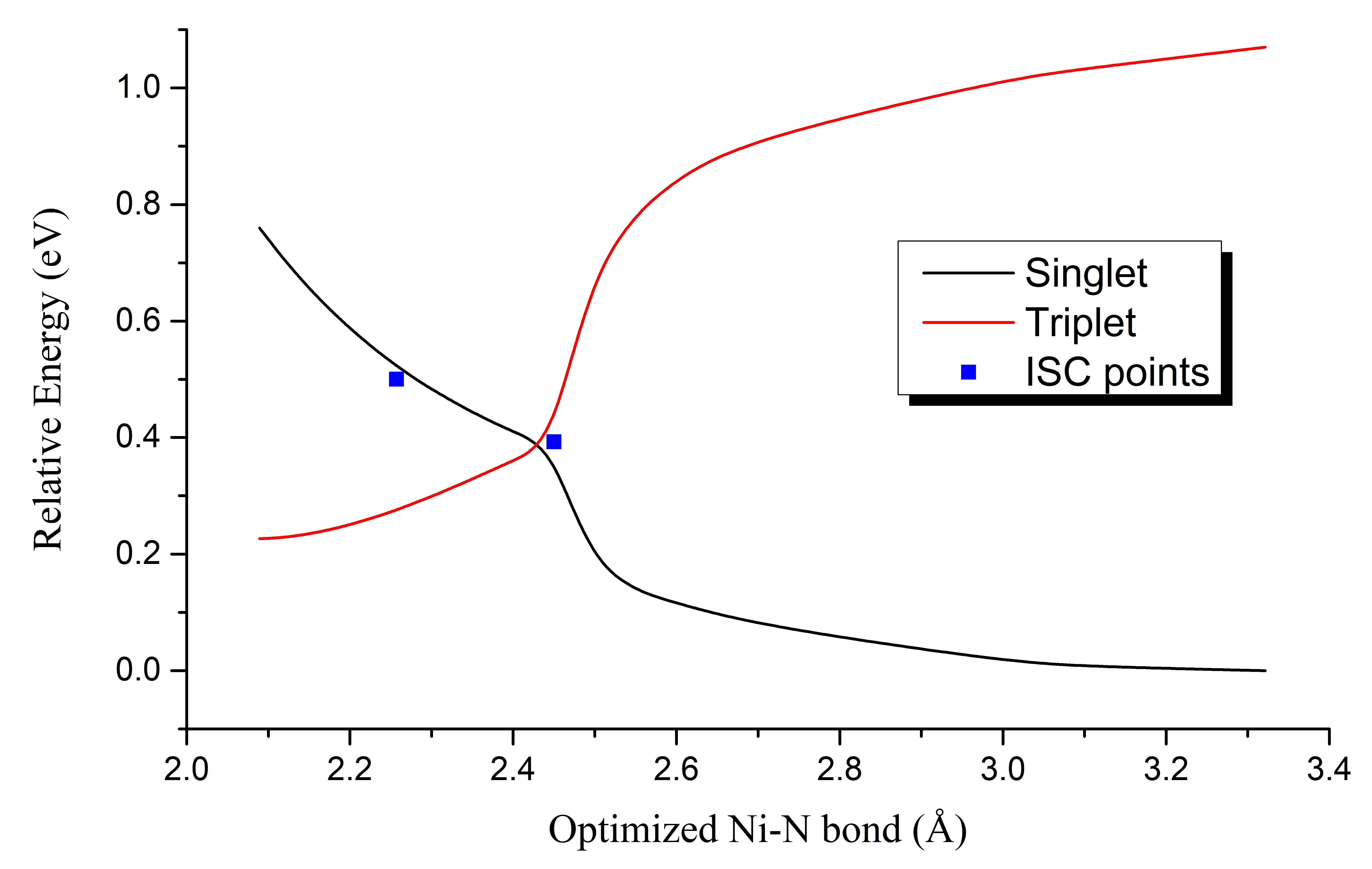
Figure 2. The variation of the relative energy for the triplet and singlet spin states and the position of the ISC points based on different d(Ni···N) distance.
It can be observed that the lowest ISC point is almost overlaps with the crossing point of the "Minimum pathway" of the d(Ni···N) distance. This means that the elongation along the minimal pathway of the d(Ni···N) distance from its starting geometry defined by the equilibrium triplet spin state geometry as well as compression along the minimal pathway of the same d(Ni···N) distance from its starting geometry defined by the equilibrium singlet spin state geometry will lead us to the same ISC point. There is also another ISC point along the minimal pathway of the d(Ni···N) distance in singlet spin state configuration but other geometrical parameters, like, d(Ni···N) distance from the porphyrine ring must changes significantly in order to reach this ISC point. Not only the localization of the intersystem crossing point is important but also the spin-orbit coupling (SOC) calculated within the ISC geometry configuration. Accordingly, we have used the MolSOC software in order to compute the different SOC values along the porphyrine planar and perpendicular directions. The SOC values are: 17.869 cm-1 in the porphyrine plane and 5.074 cm-1 in the perpendicular direction to this plane. These SCO values can be considered as strong couplings and we can conclude that once the ISC geometry is reached the spin flipping take place with a high probability.
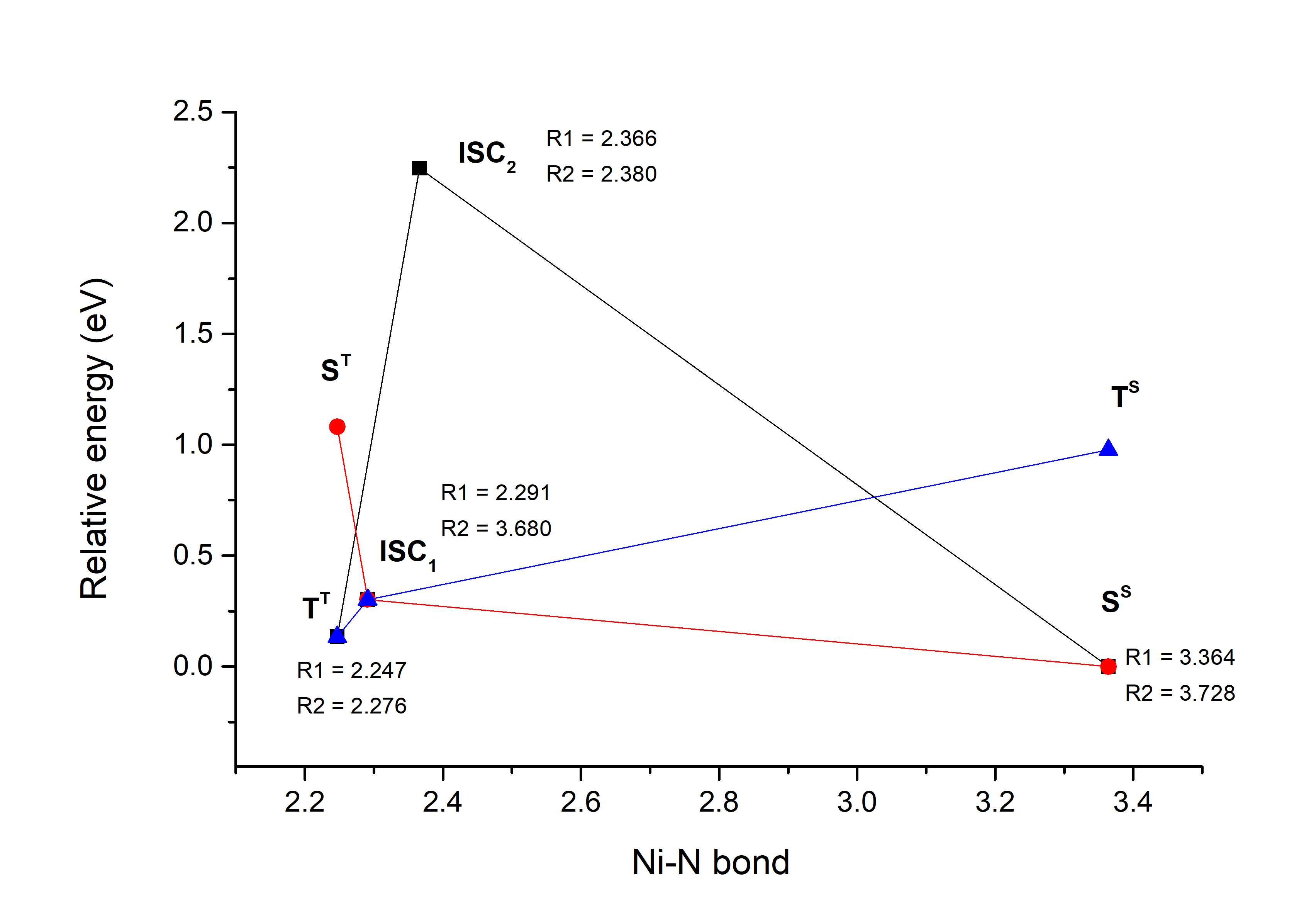
Ni-P-biAP. This molecular complex contains two azopiridines mobile fragment which can creates not one but two further ligand bonds with the central Ni atom, making in this way an octahedral (six-coordonated) metal ligand structure. Using the homemade developed PyISC code we have located two ISC geometries, which two different topological characteristics. The first one (ISC1), with low relative energy (Δ=0.302 eV) is an antisymmetric configuration where one of the ligands made by the azopiridine fragments is moved away from the central Ni atom, keeping a pyramidal-planar ligand configuration (R1=2.291 Å and R2=3.680 Å). The second ISC2 geometry is much higher in its relative energy (Δ=2.247 eV), but shows a near-symmetric ligand configuration where both ligand bonds formed by the azopiridine fragments are almost similarly changed (R1=2.366 Å and R2=3.380 Å). For the definition of the R1 and R2 ligand bonds see Figure 3a, and for the relative energy for the triplet and singlet spin states and the position of the ISC points see Figure 3b. The SOC values for the ISC1 geometry are: 12.780 cm-1 in the porphyrine plane and 1.165 cm-1 in the perpendicular direction to this plane, while for the ISC2 geometry are: 10.590 cm-1 in the porphyrine plane and 1.959 cm-1 in the perpendicular direction to this plane.
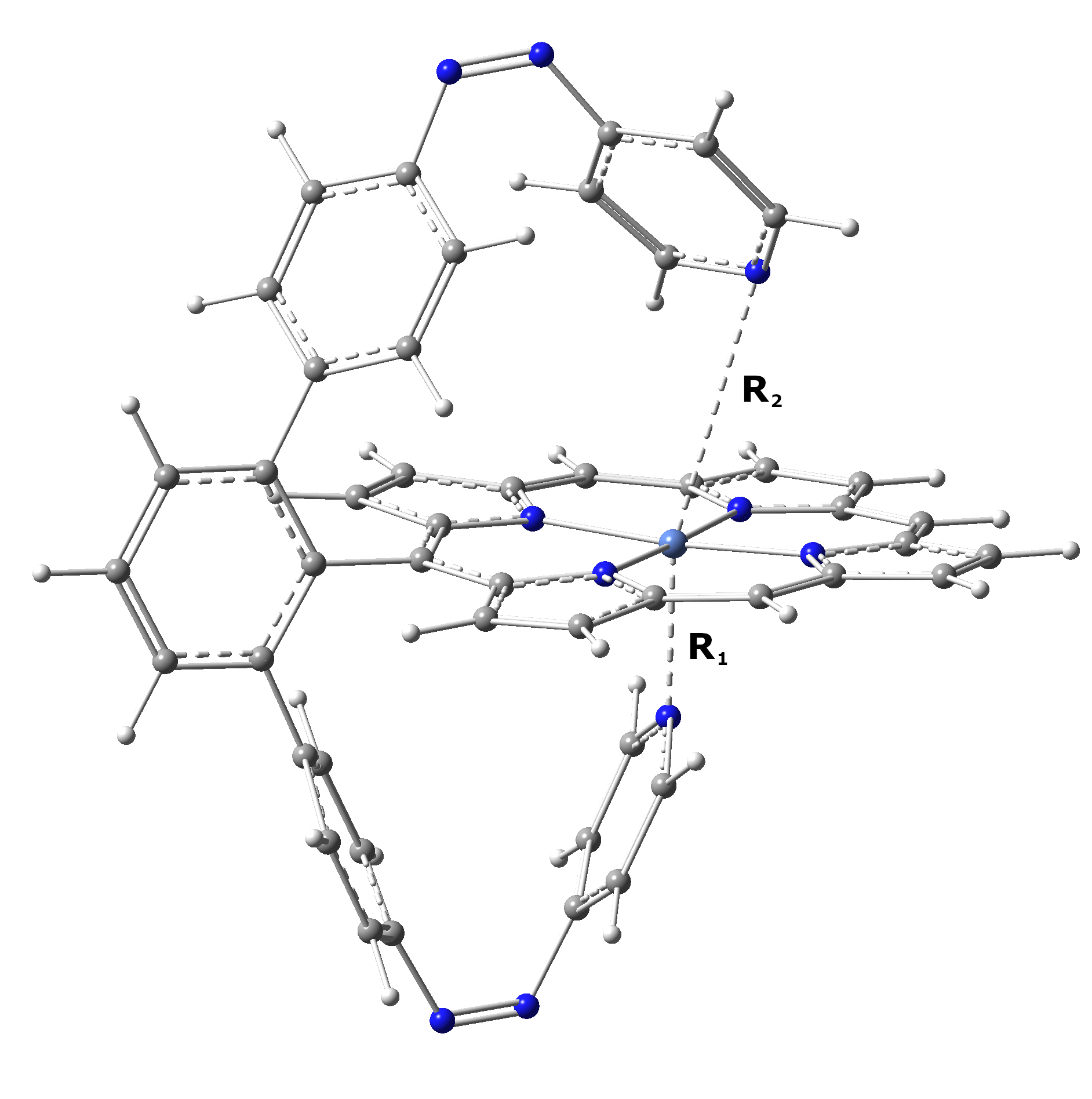
(a)
(b)
Figure 3. (a) The geometry configuration of the Ni-P-biAP molecular complex; (b) The energetic scheme of the equilibrium geometries for singlet and triplet spin states as well as the geometries of the ISC point for the Ni-P-biAP molecular complex.
One can conclude that the triplet - singlet spin transition involves three different metal-ligand coordination configuration. The octahedral triplet metal-ligand coordination complex changes its geometry to the pyramidal-planar ligand form in its transition state configuration characterized by the ISC1 geometry and finally changes in the singlet spin state configuration taking the square-planar metal-ligand coordination form (See Figure 4).
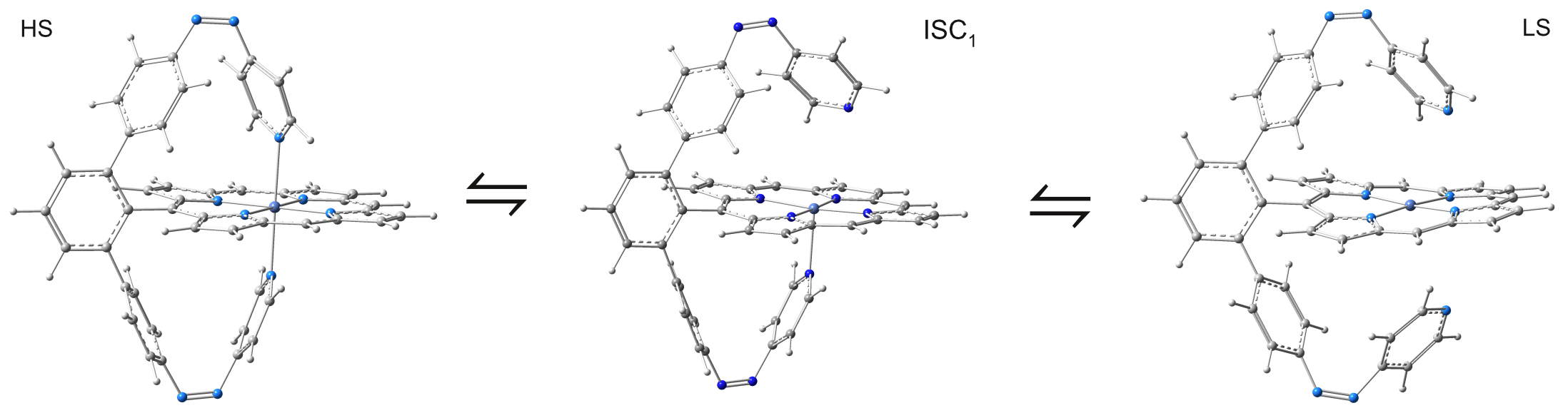
Figure 4. The variation of the metal-ligand coordination along the triplet - ISC - singlet transition.
It is also worth to mention that the spin transition mechanism where both ligand bonds formed by the azopiridine fragments are almost similarly changed is not energetically preferred.
Vertical Excitation Energies
In both cases of Ni-TPP-AP and Ni-P-biAP molecular complexes the energy barrier between the "Low-spin" and "High-spin" states defined by the ISC geometry is around 0.3 - 0.4 eV. In order to pass through the barrier more easily and in a controlled manner the electronic excitation of the complexes is considered. Accordingly, one needs to know in detail the nature of the vertical excitation energy levels for each molecular complexes considering both the LS and HS states.
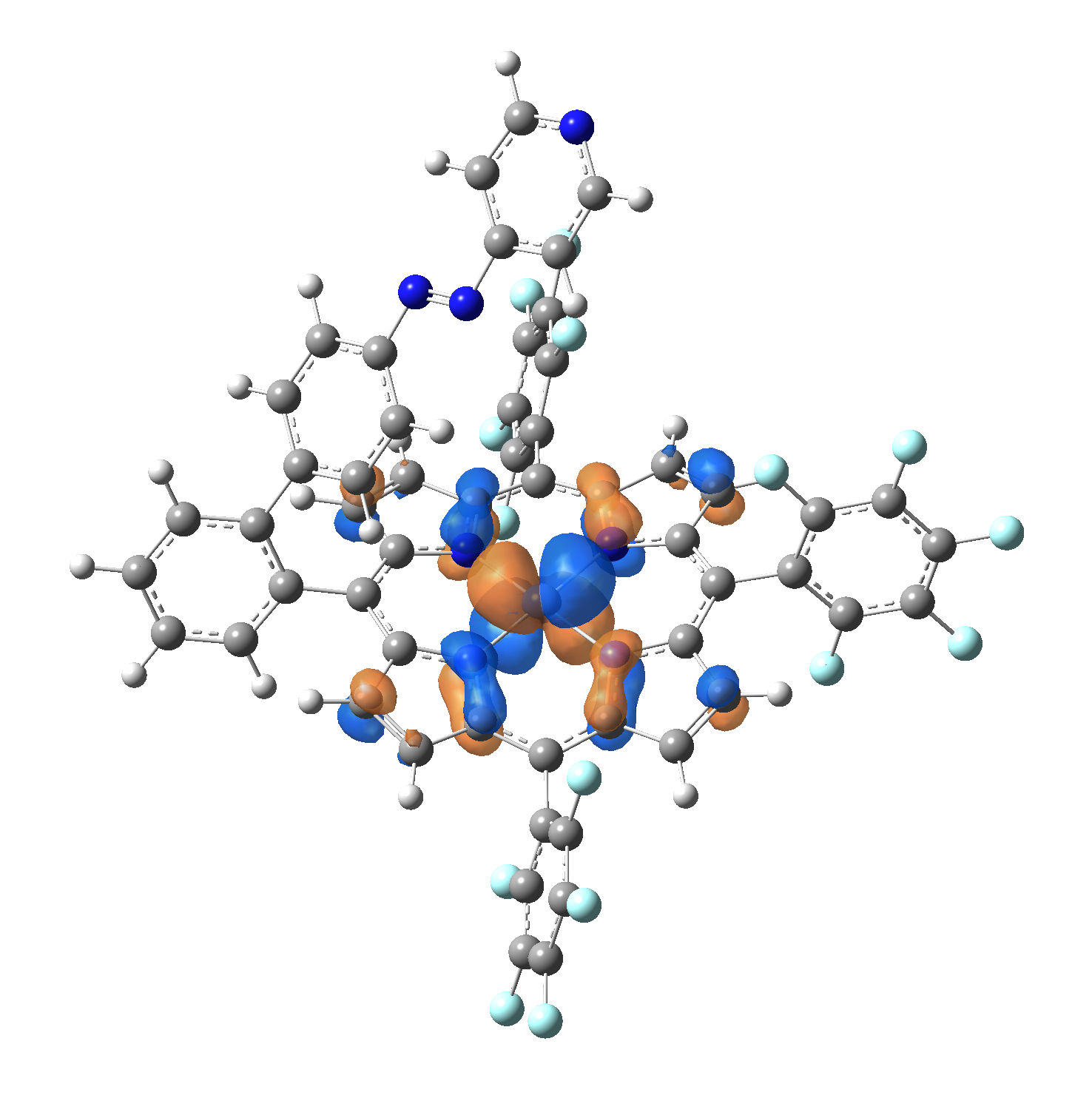
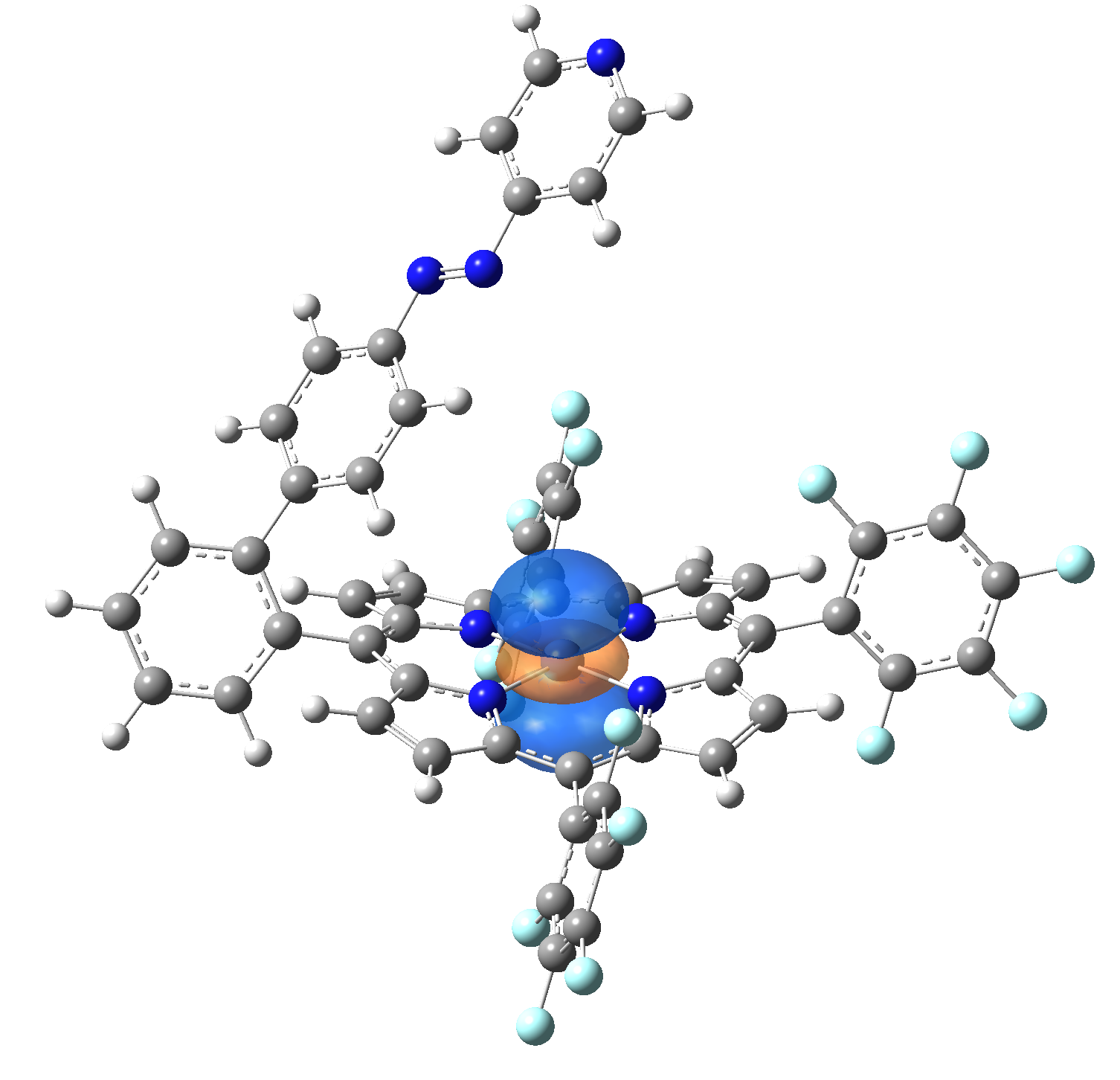
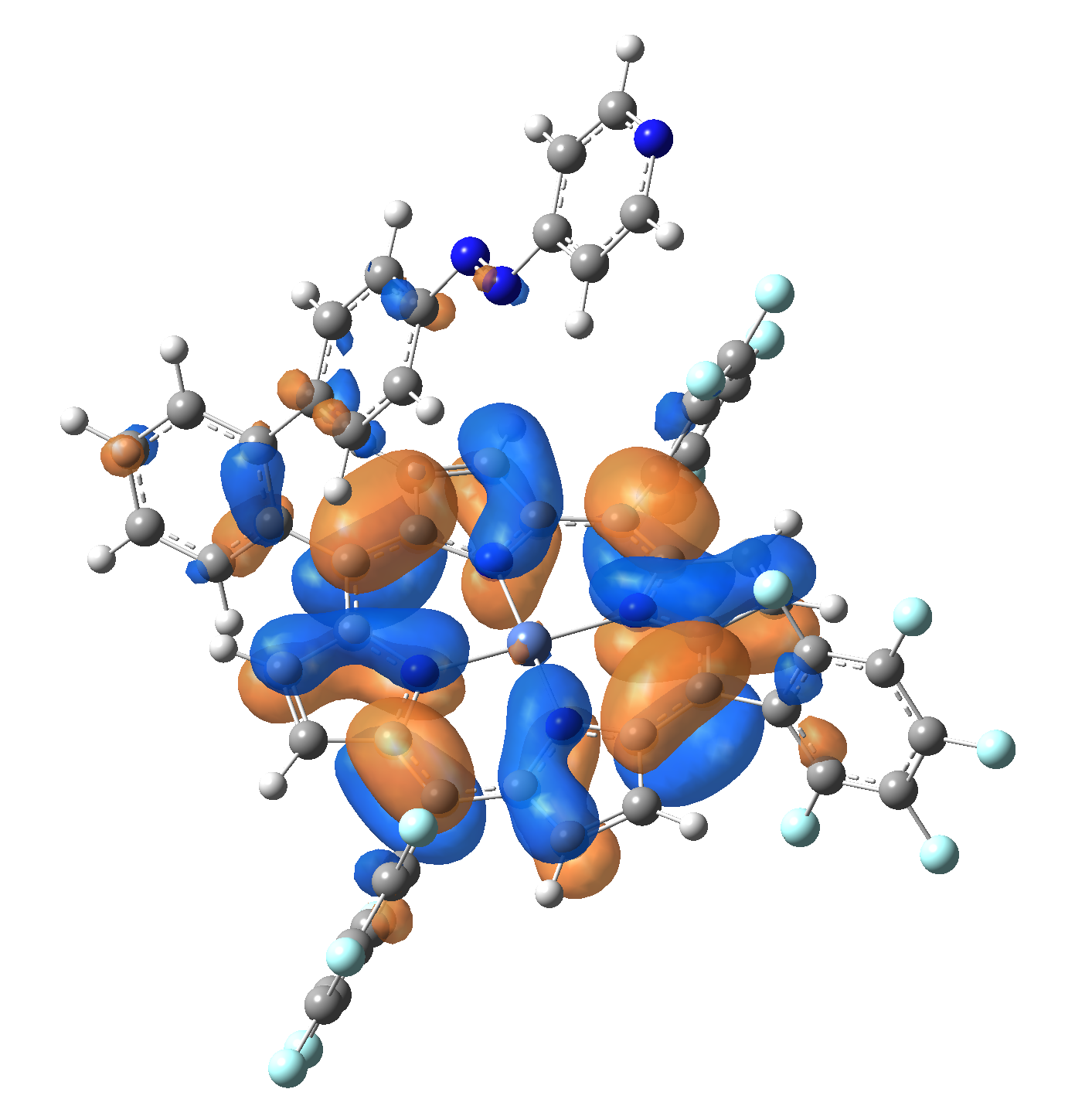
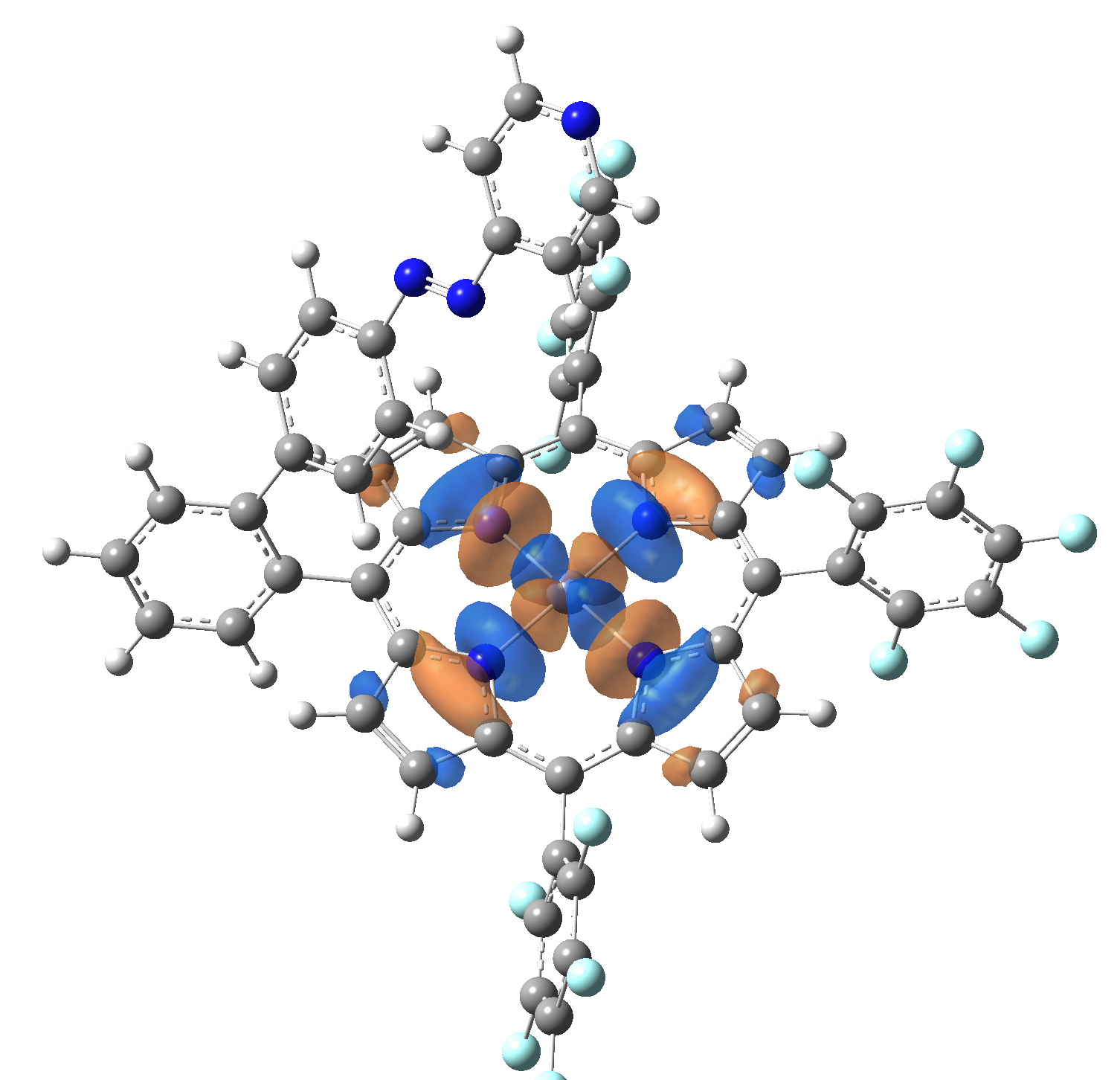
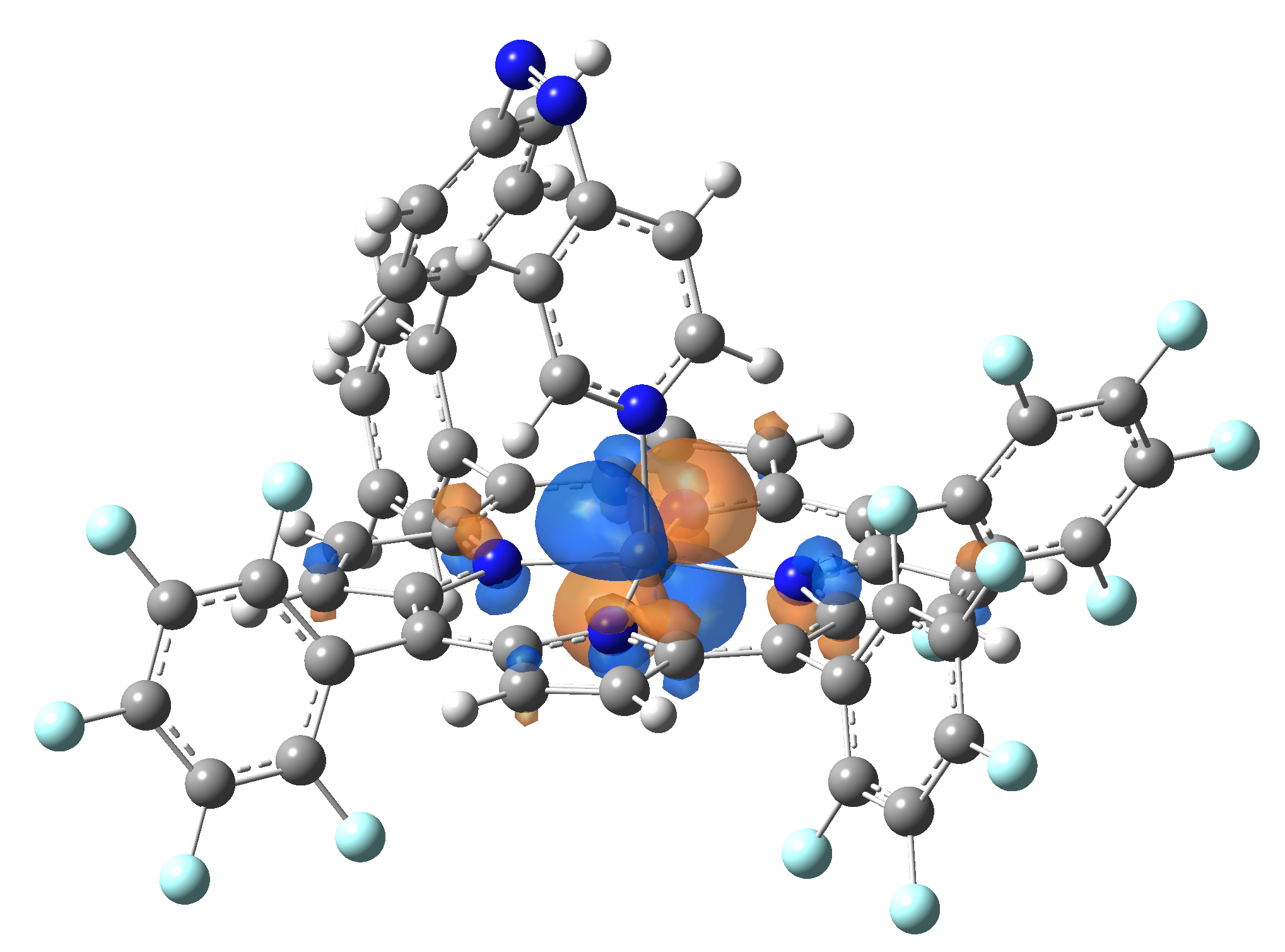
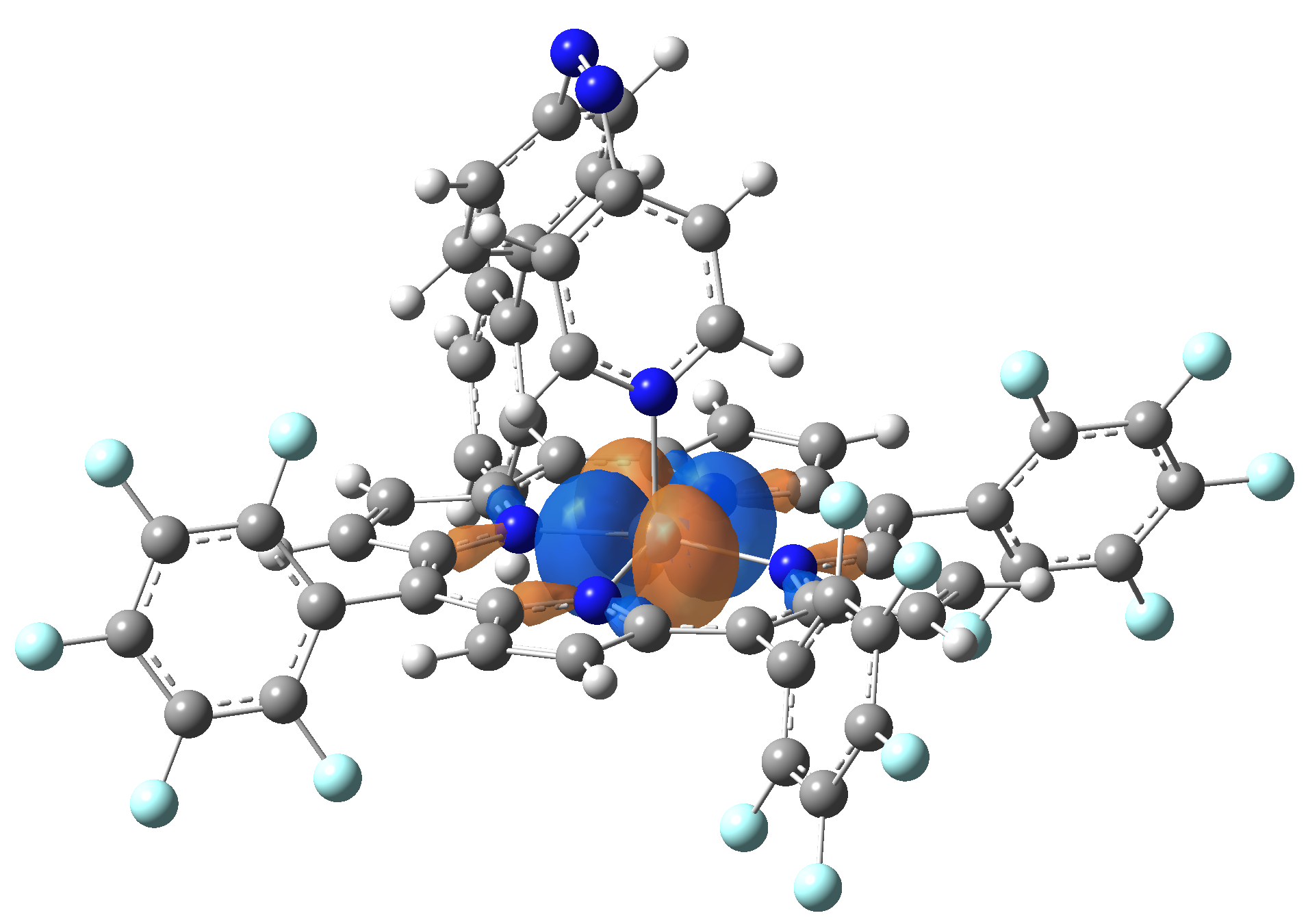
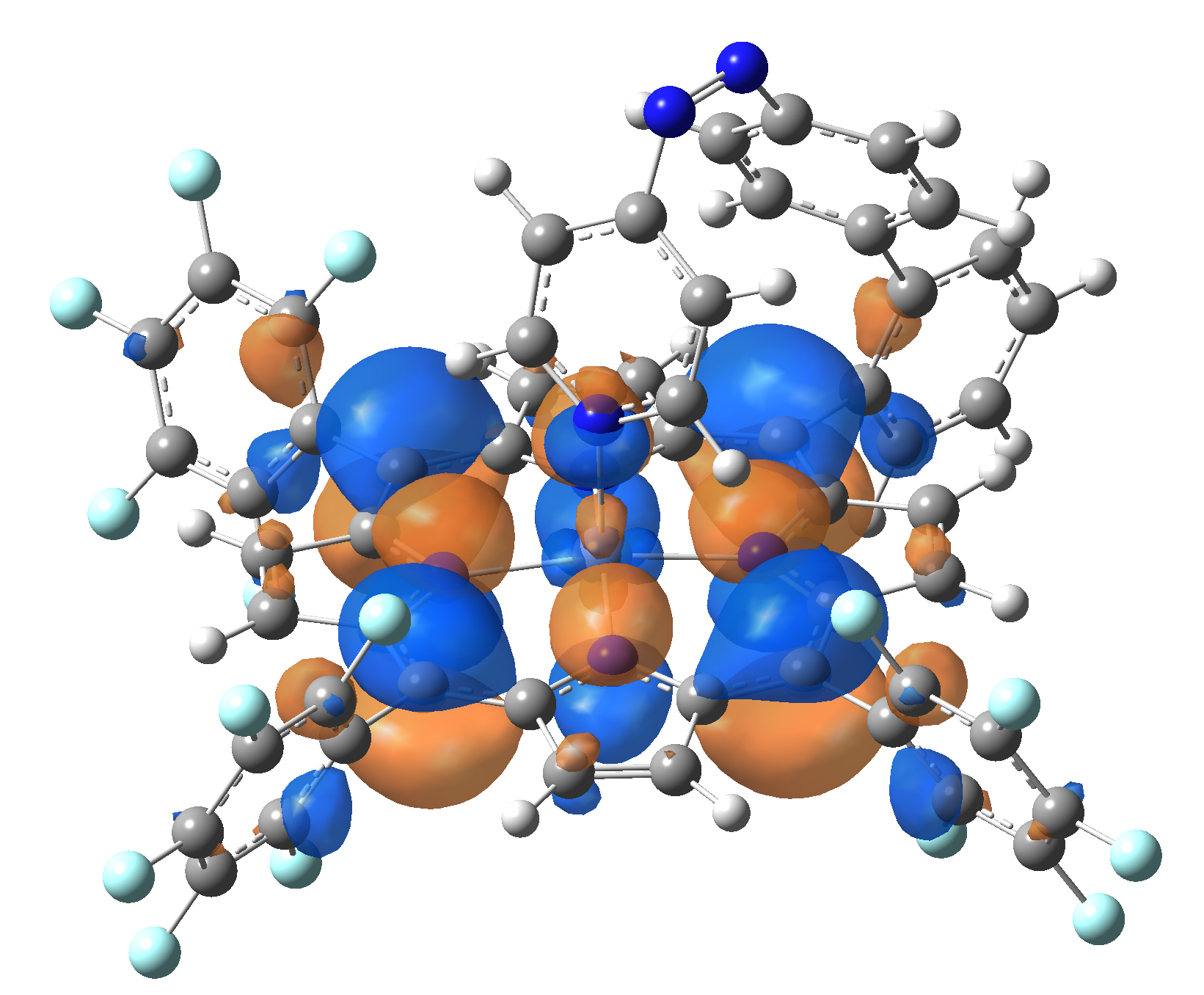
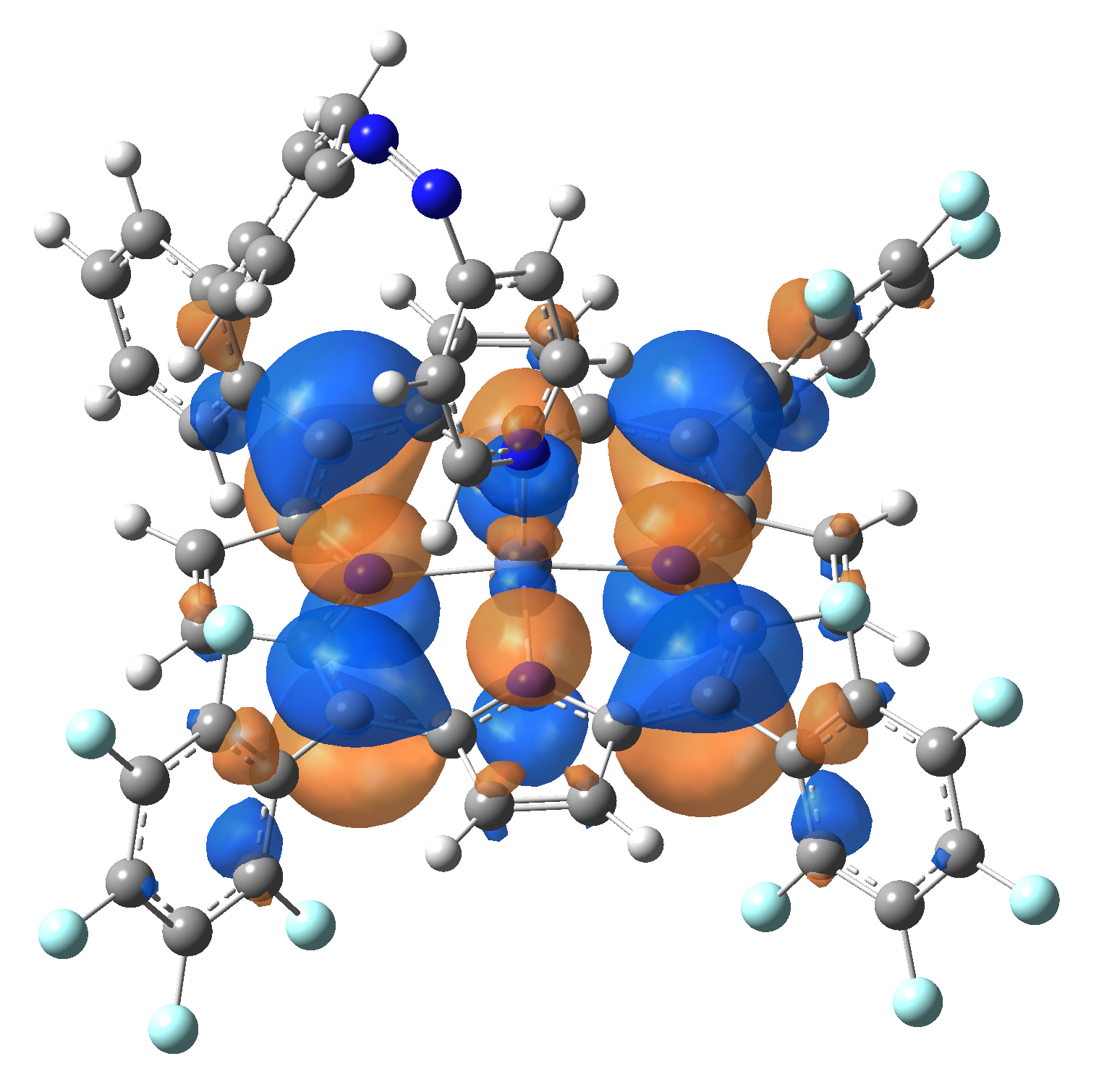
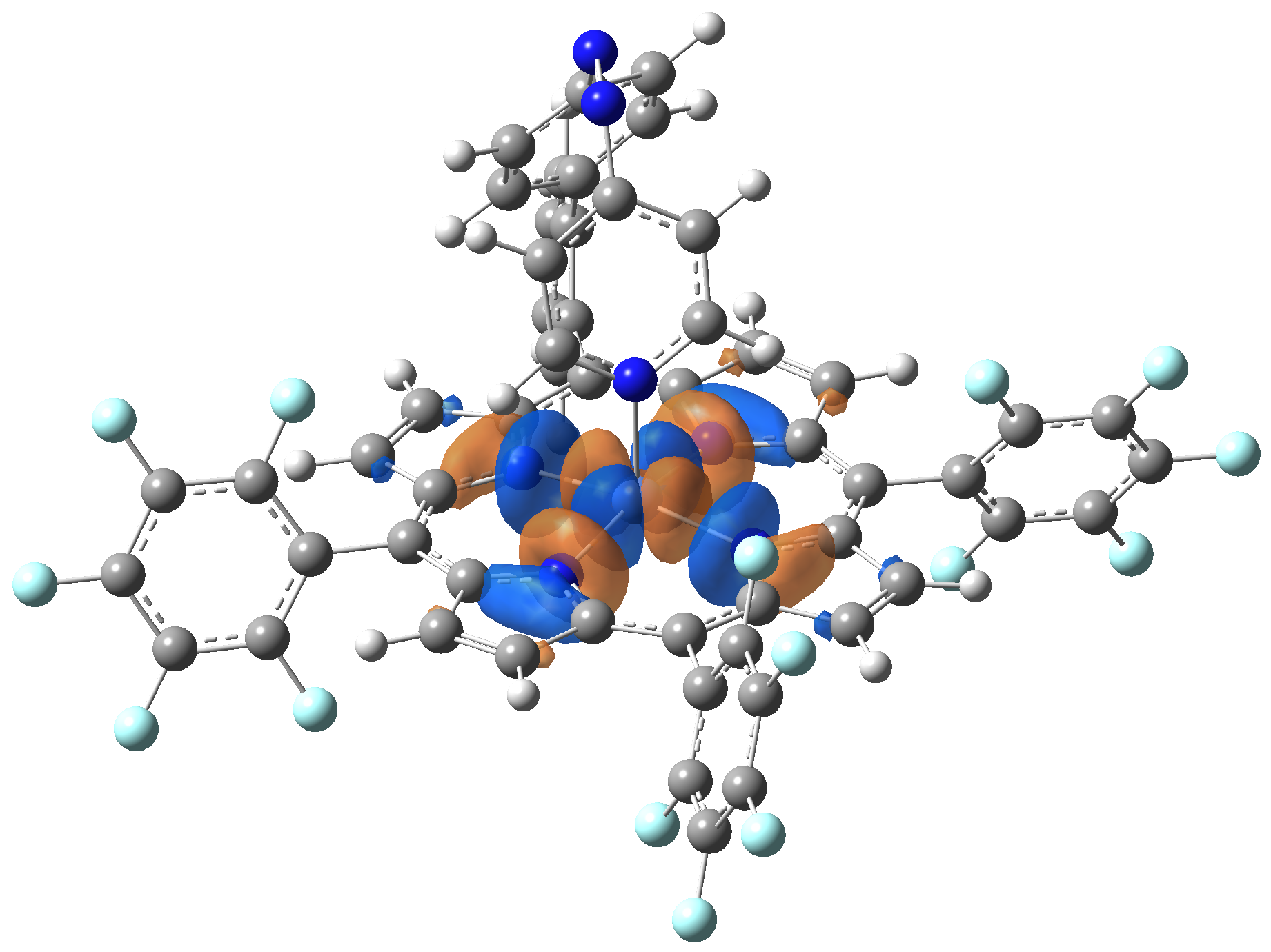
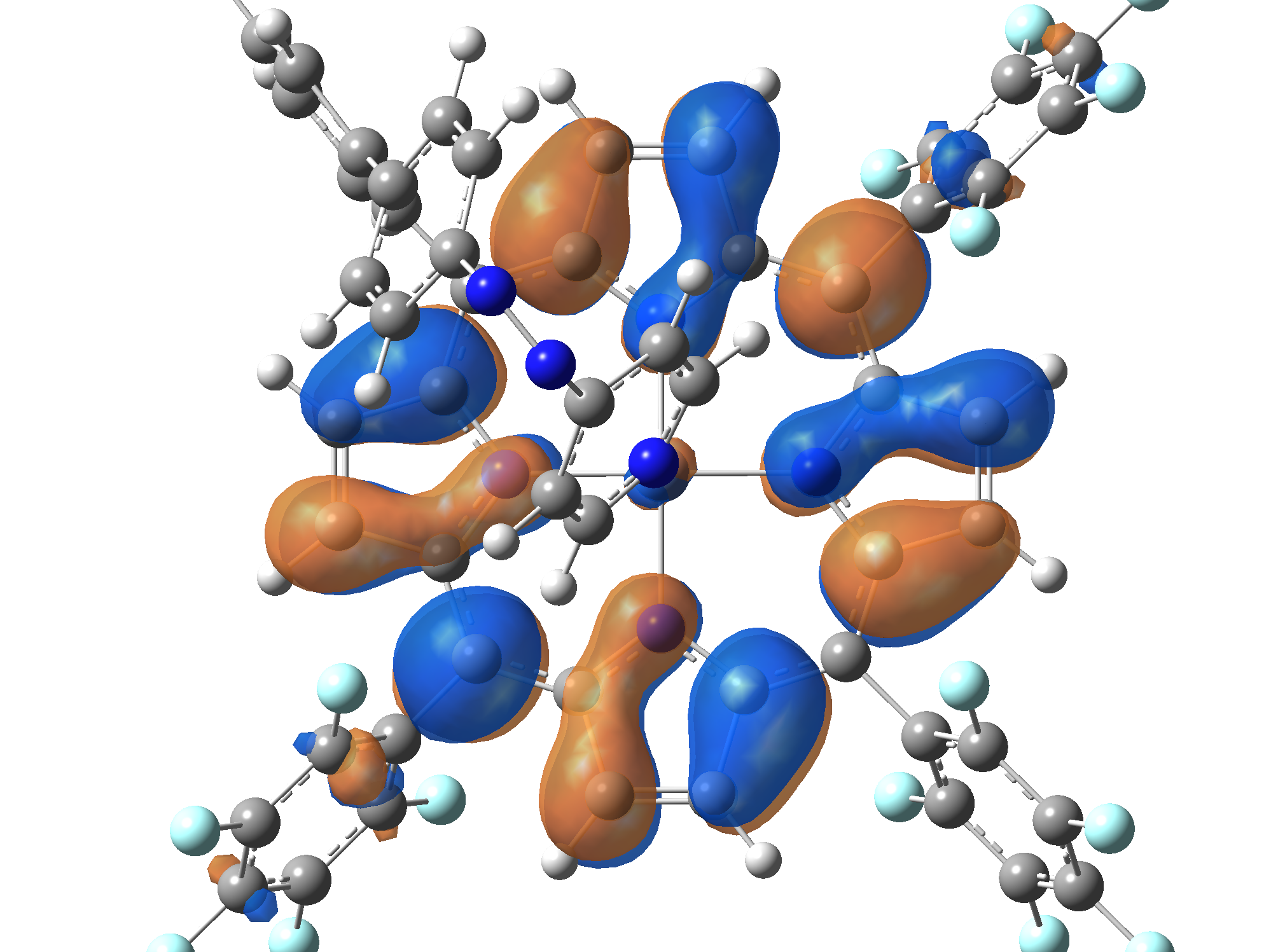
Ni-TPP-AP. The first 30 electronic excited state have been computed at MN12-SX/def2-TZVP level of theory. The band origin (the energy value of the S1 electronic state) of the vertical excitation for the singlet spin state starts at 528.71 nm, but the first active excited state (with nonzero oscillator strength) is the S4 electronic state (499.99 nm). The band origin of the vertical excitation for the triplet spin state starts at 1068.93 nm, and the first active state is the S5 electronic state (632.46 nm). The most eloquent characteristic of the electronic excited state is the so-called Natural Transition Orbitals (NTO), namely, the density changes induced by the electronic transition from the ground to the given excited electronic state. The energy and oscillator strength as well as the Highest Occupied Molecular Orbitals (HOMO) and the Lowest Unoccupied Molecular Orbitals (LUMO) for the first 5 electronic transition with singlet spin state is shown in Table 1
| Exc. State | S1 | S2 | S3 | S4 | S5 |
|---|---|---|---|---|---|
| Energy | 2.3450 eV | 2.3592 eV | 2.3646 eV | 2.4797 eV | 2.4935 eV |
| 528.71 nm | 525.53 nm | 524.32 nm | 499.99 nm | 497.23 nm | |
| Osc. strength | f=0.0000 | f=0.0000 | f=0.0000 | f=0.0005 | f=0.0029 |
| HOMO | 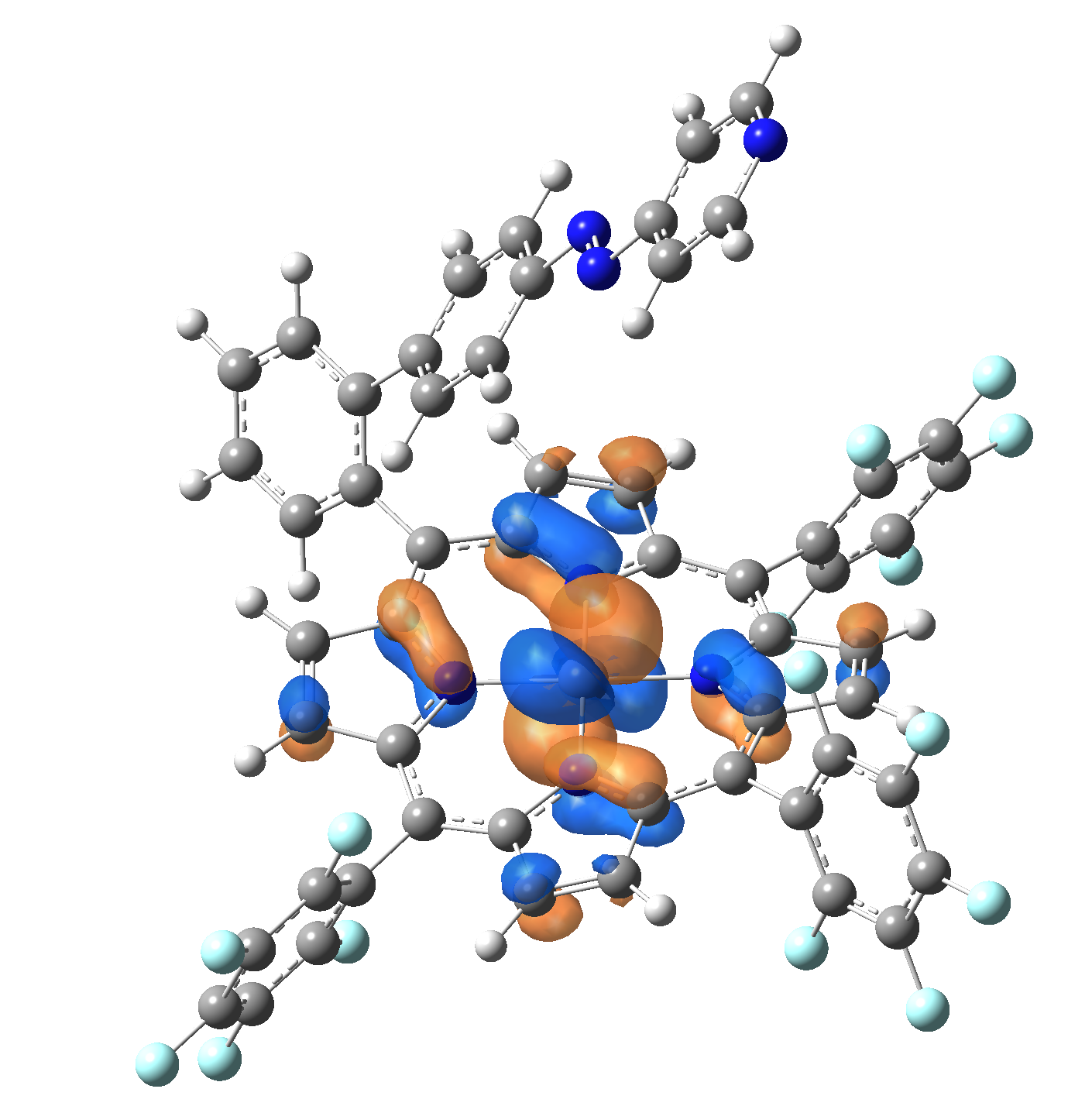 |
 |
 |
 |
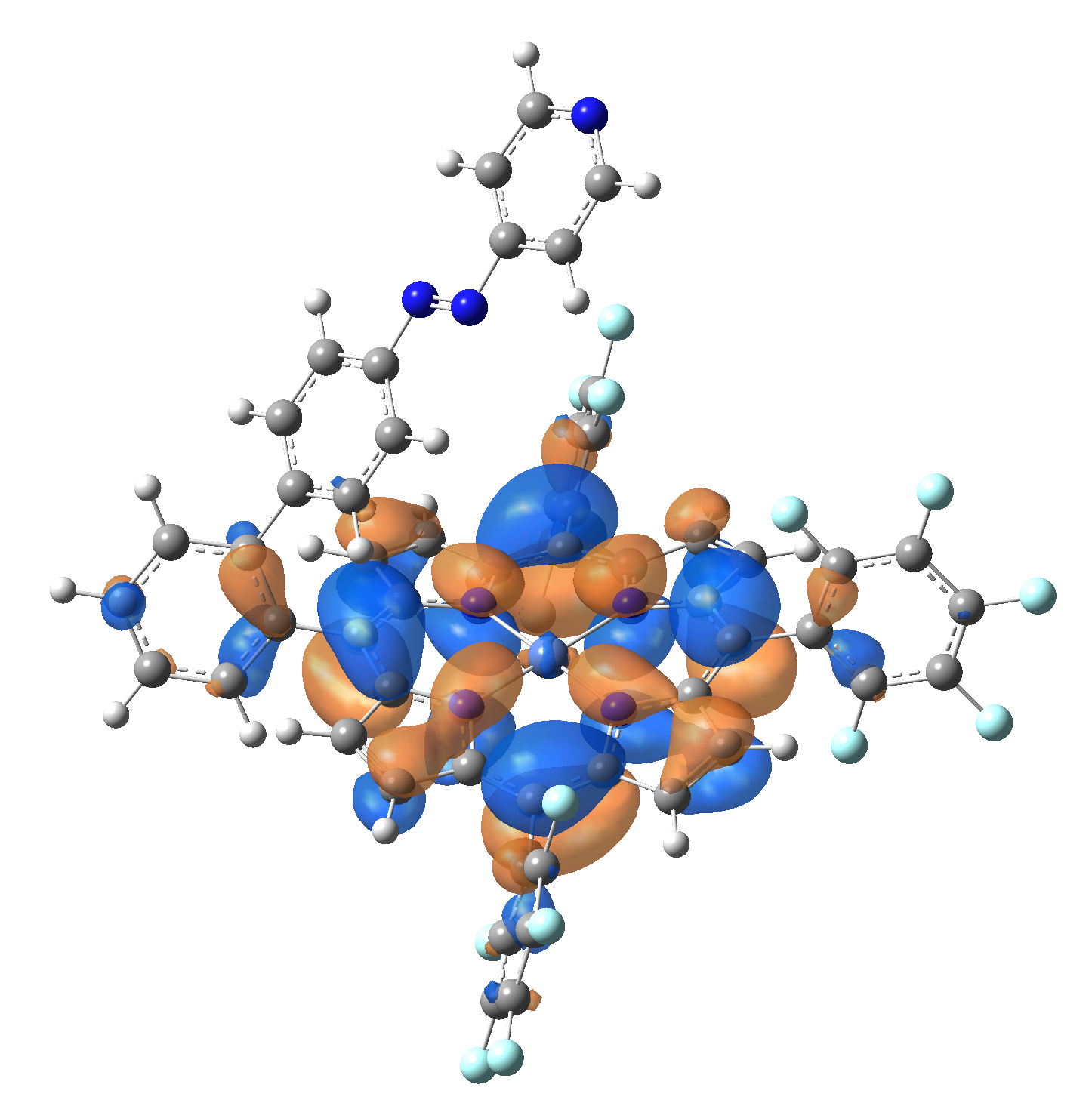 |
| LUMO | 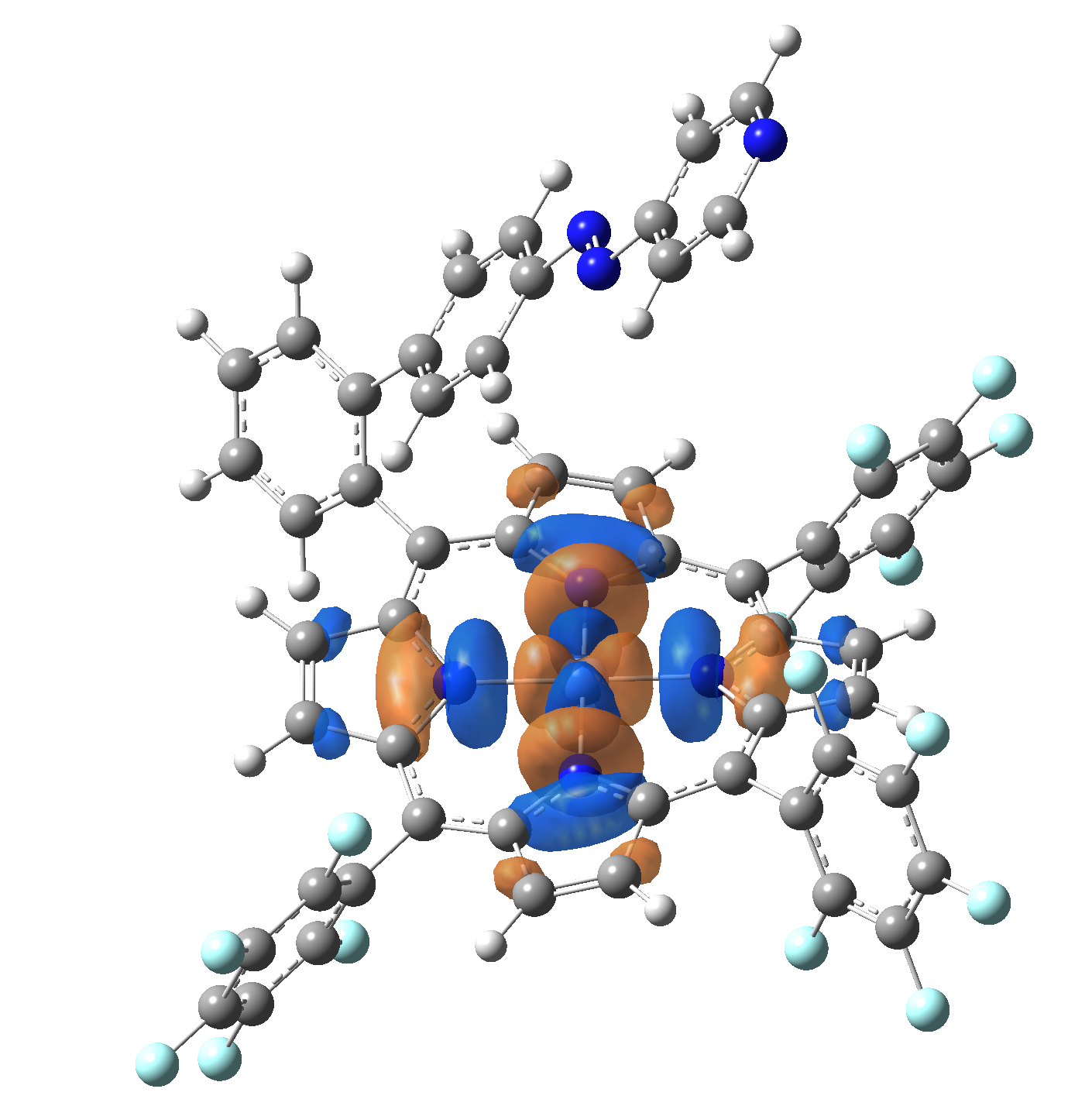 |
 |
 |
 |
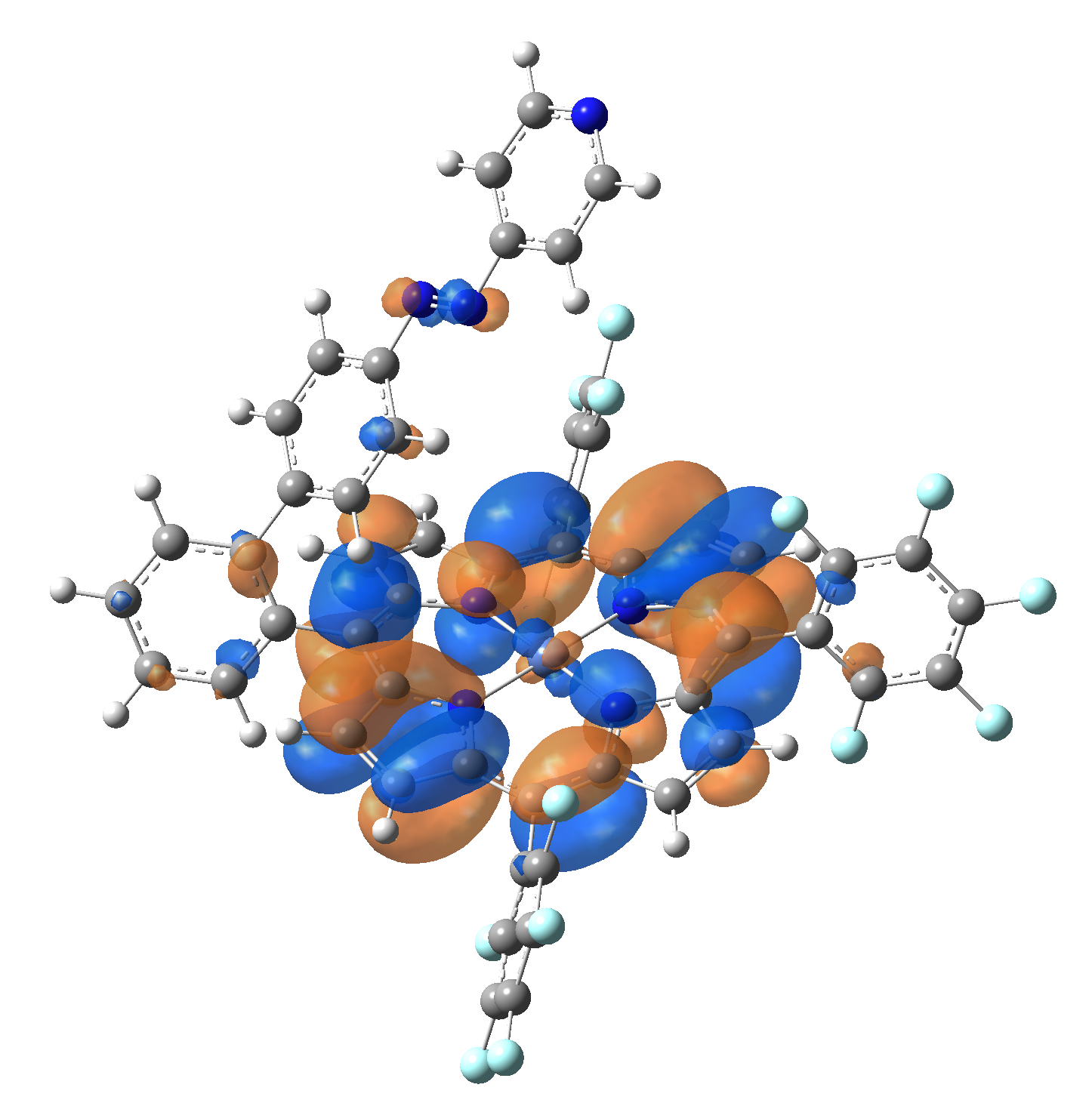 |
Table 1. The energy and oscillator strength as well as the HOMO and the LUMO NTO orbitals for the first 5 electronic transition with singlet spin state of the Ni-TPP-AP molecular complex.
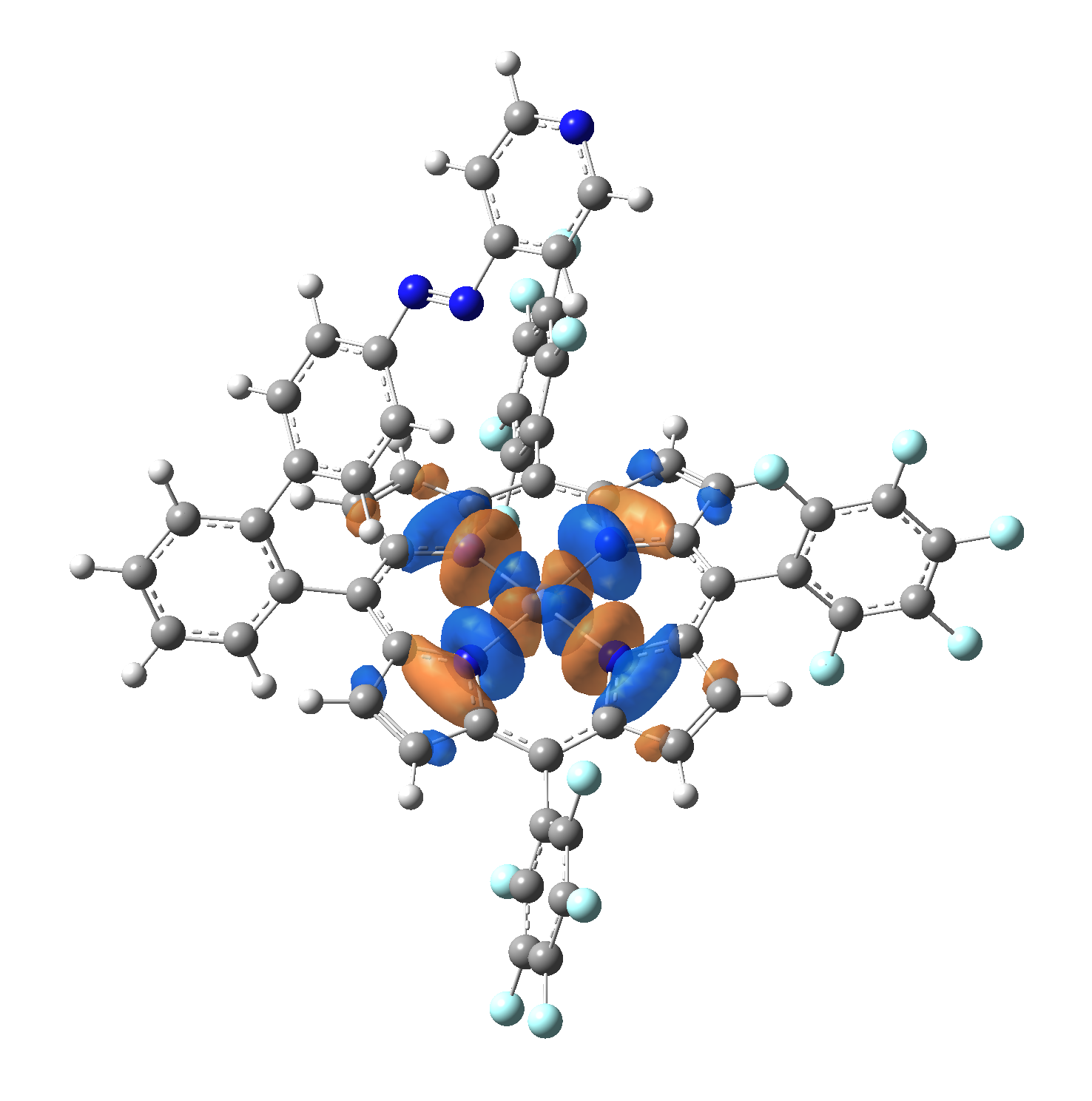
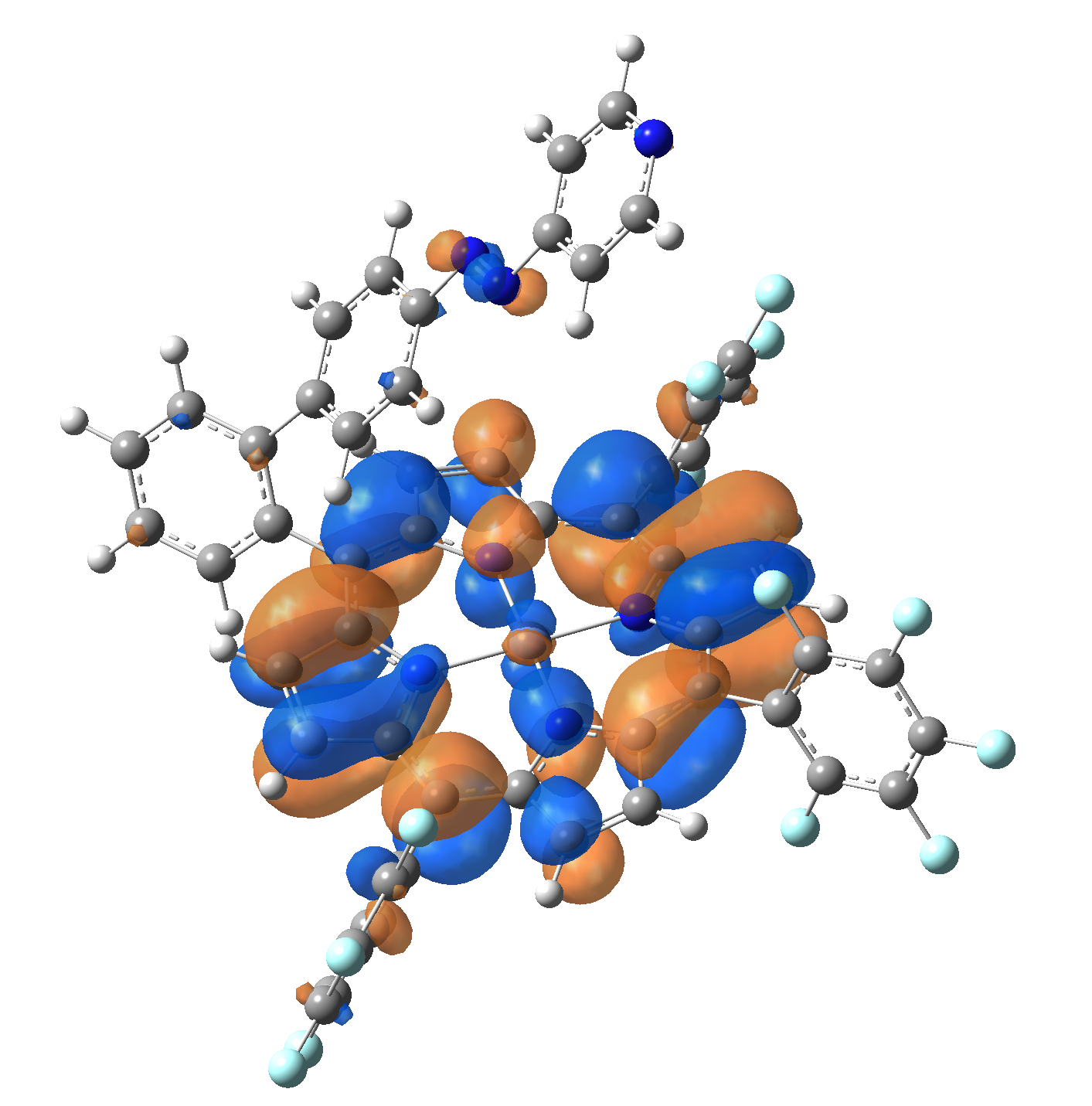
The energy and oscillator strength as well as the Highest Occupied Molecular Orbitals (HOMO) and the Lowest Unoccupied Molecular Orbitals (LUMO) for the first 10 electronic transition with triplet spin state is shown in Table 2
| Exc. State | T1 | T2 | T3 | T4 | T5 |
|---|---|---|---|---|---|
| Energy | 1.1599 eV | 1.1922 eV | 1.8194 eV | 1.8989 eV | 1.9603 eV |
| 1068.93 nm | 1039.95 nm | 681.45 nm | 652.93 nm | 632.46 nm | |
| Osc. strength | f=0.0000 | f=0.0000 | f=0.0000 | f=0.0001 | f=0.0003 |
| HOMO | 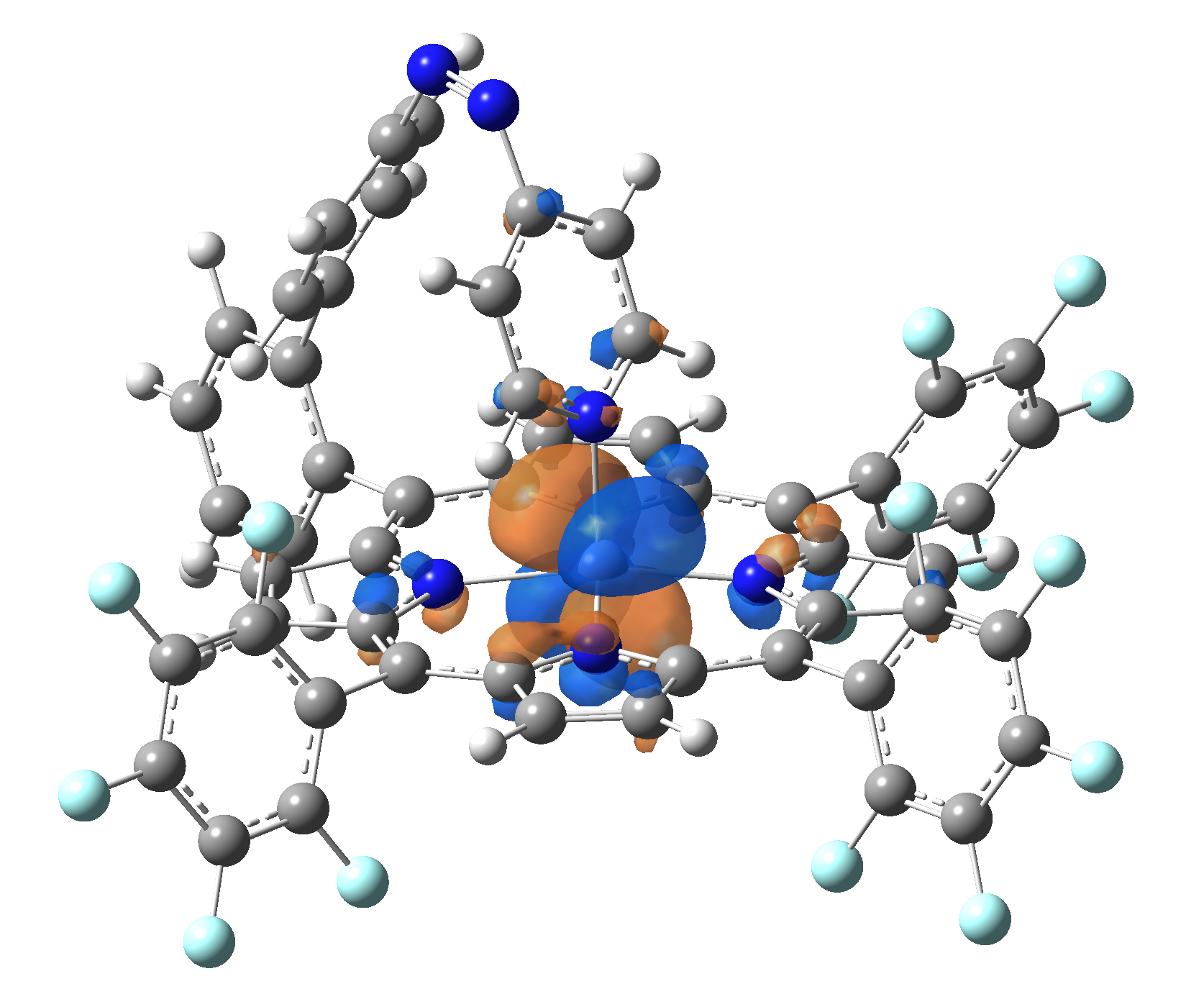 |
 |
 |
 |
 |
| LUMO | 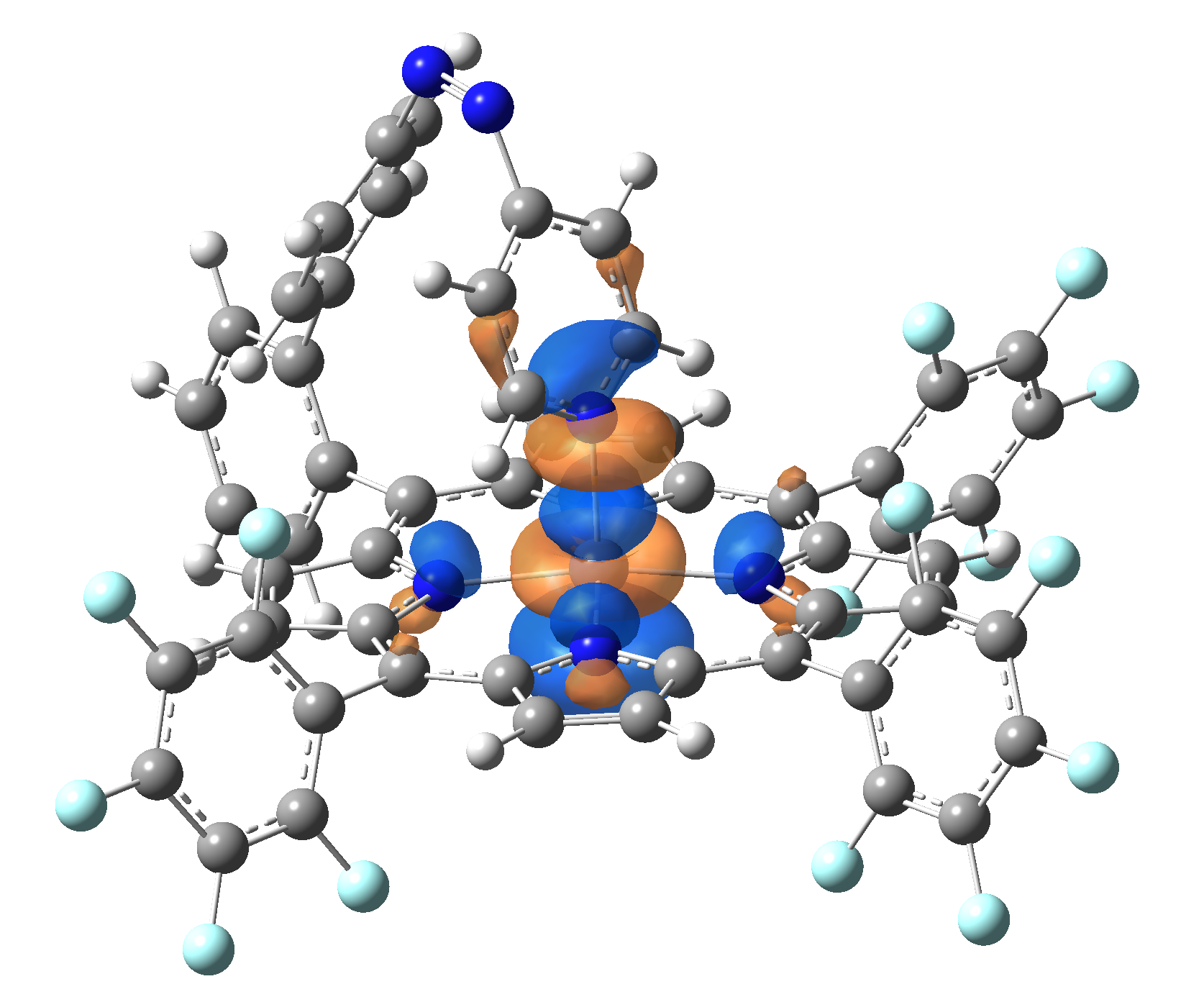 |
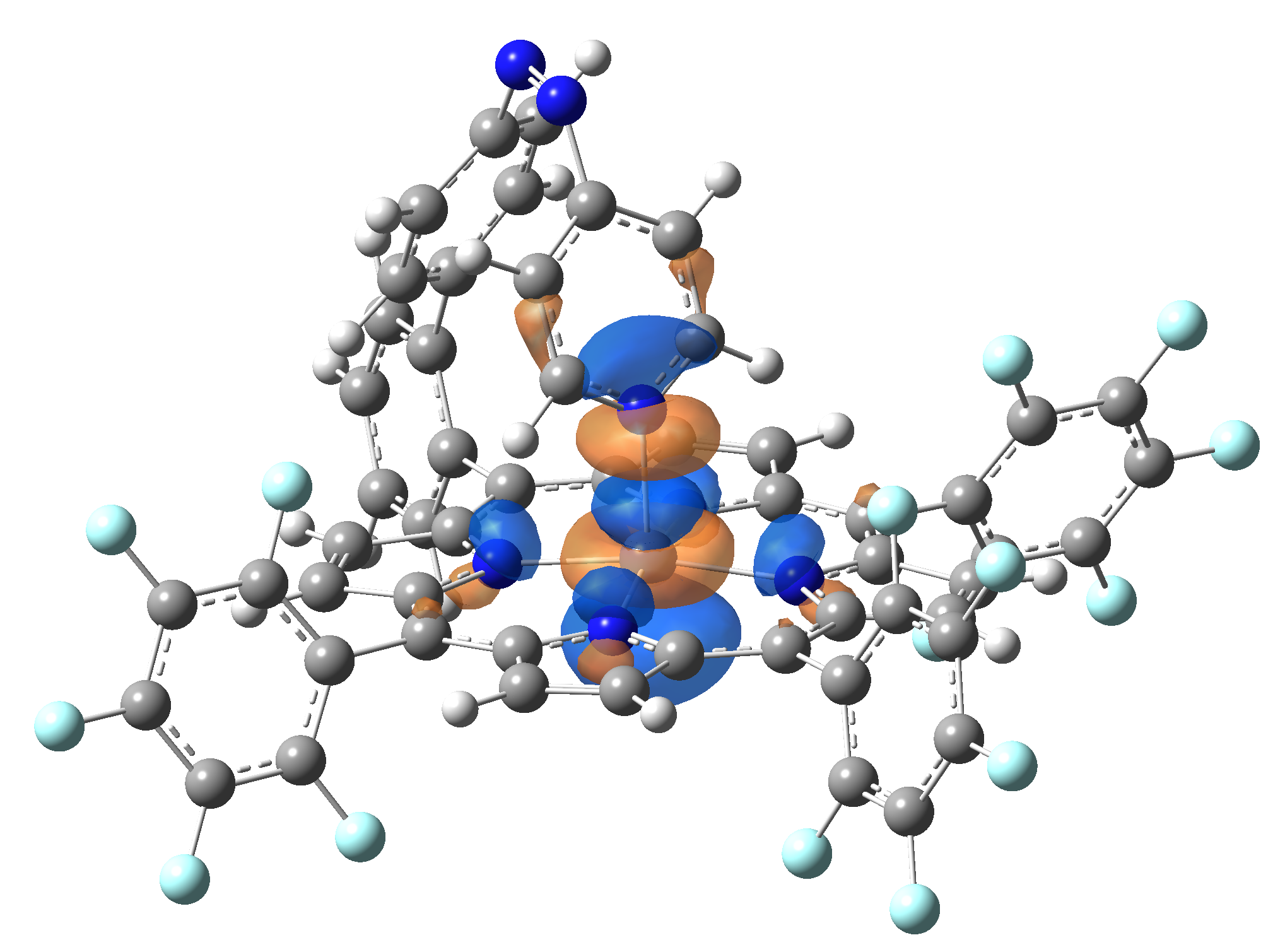 |
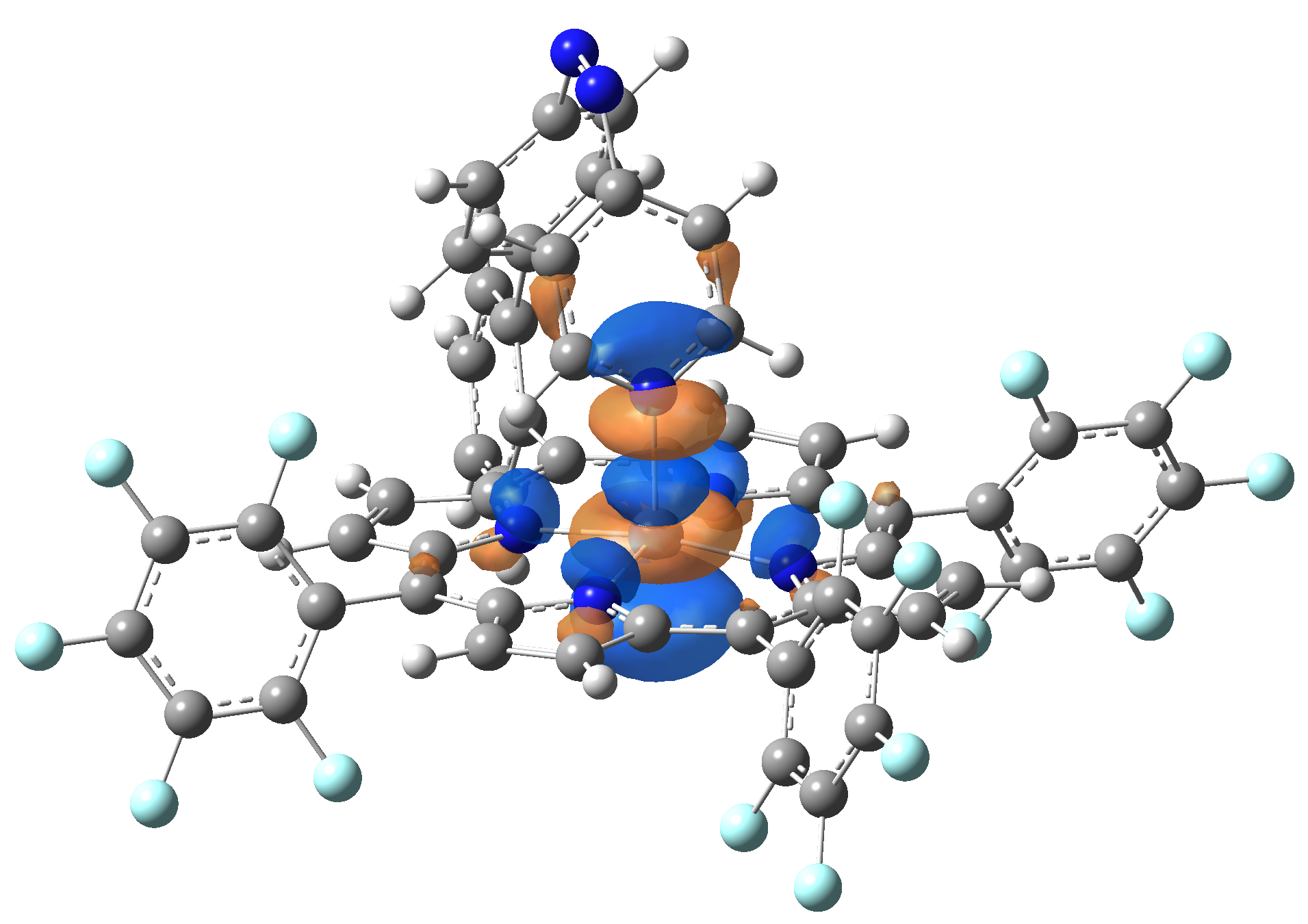 |
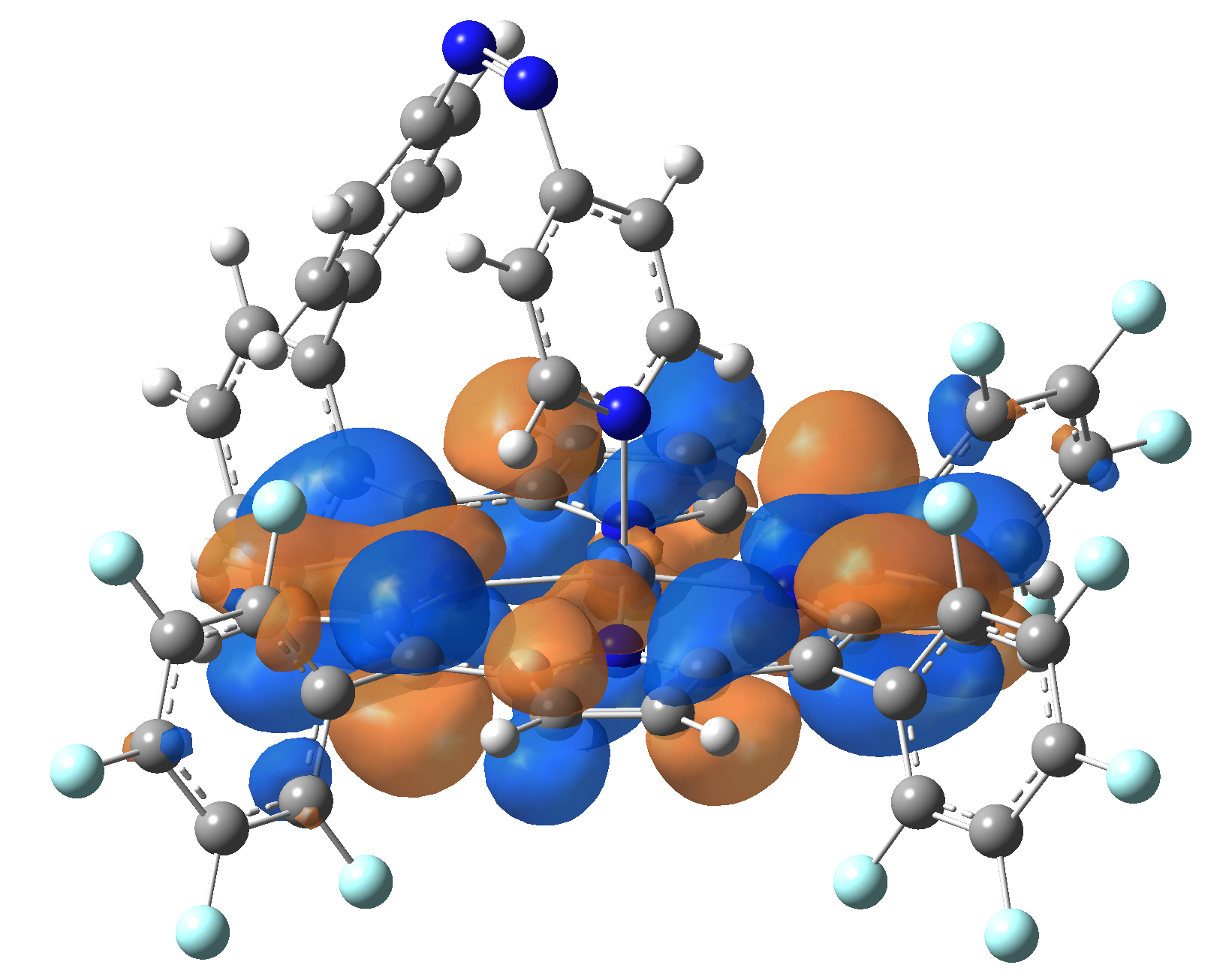 |
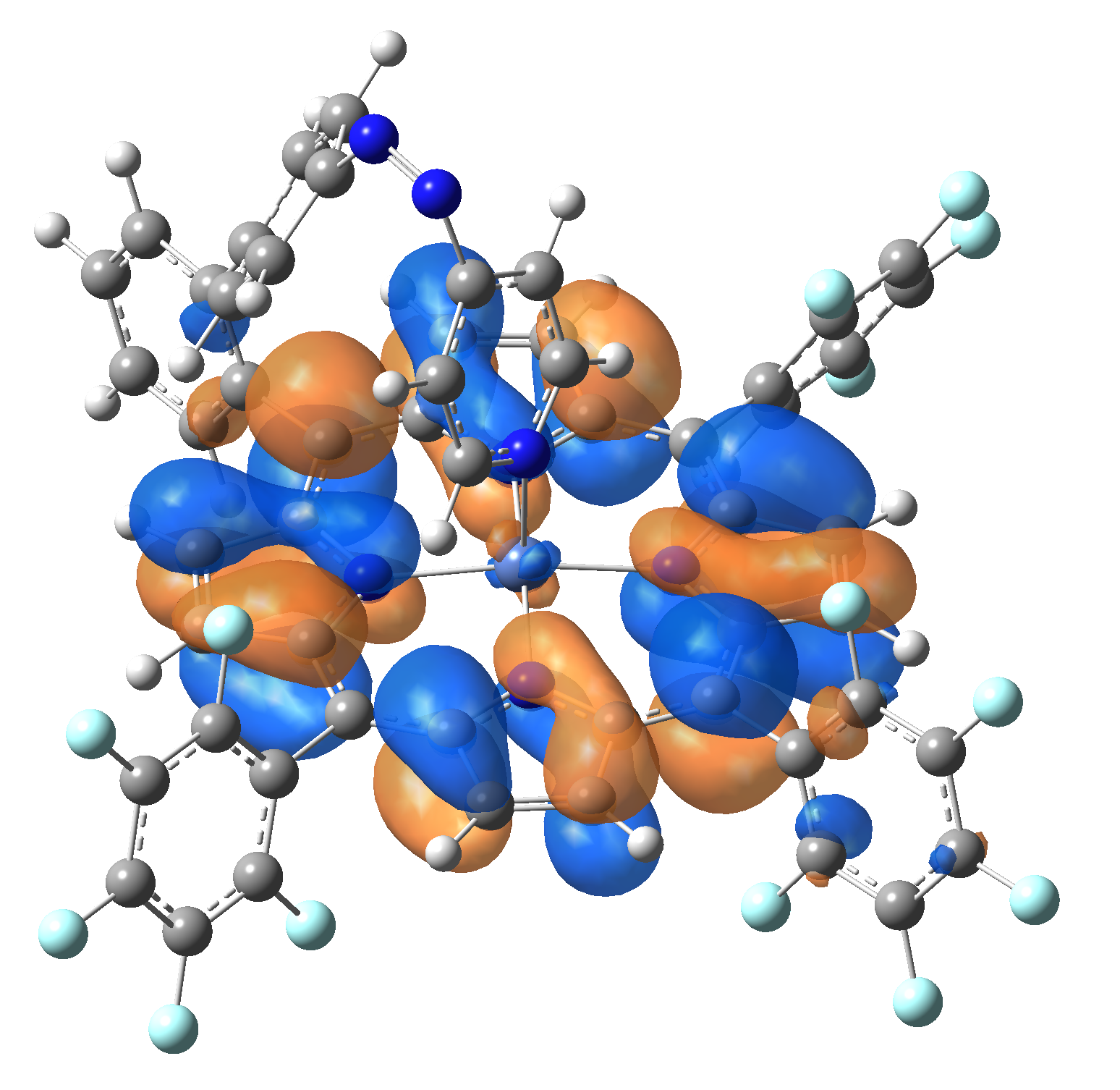 |
| Exc. State | T6 | T7 | T8 | T9 | T10 |
| Energy | 2.0584 eV | 2.1355 eV | 2.3230 eV | 2.3608 eV | 2.3925 eV |
| 602.34 nm | 580.59 nm | 533.72 nm | 525.18 nm | 518.21 nm | |
| Osc. strength | f=0.0003 | f=0.0001 | f=0.0000 | f=0.0066 | f=0.0004 |
| HOMO | 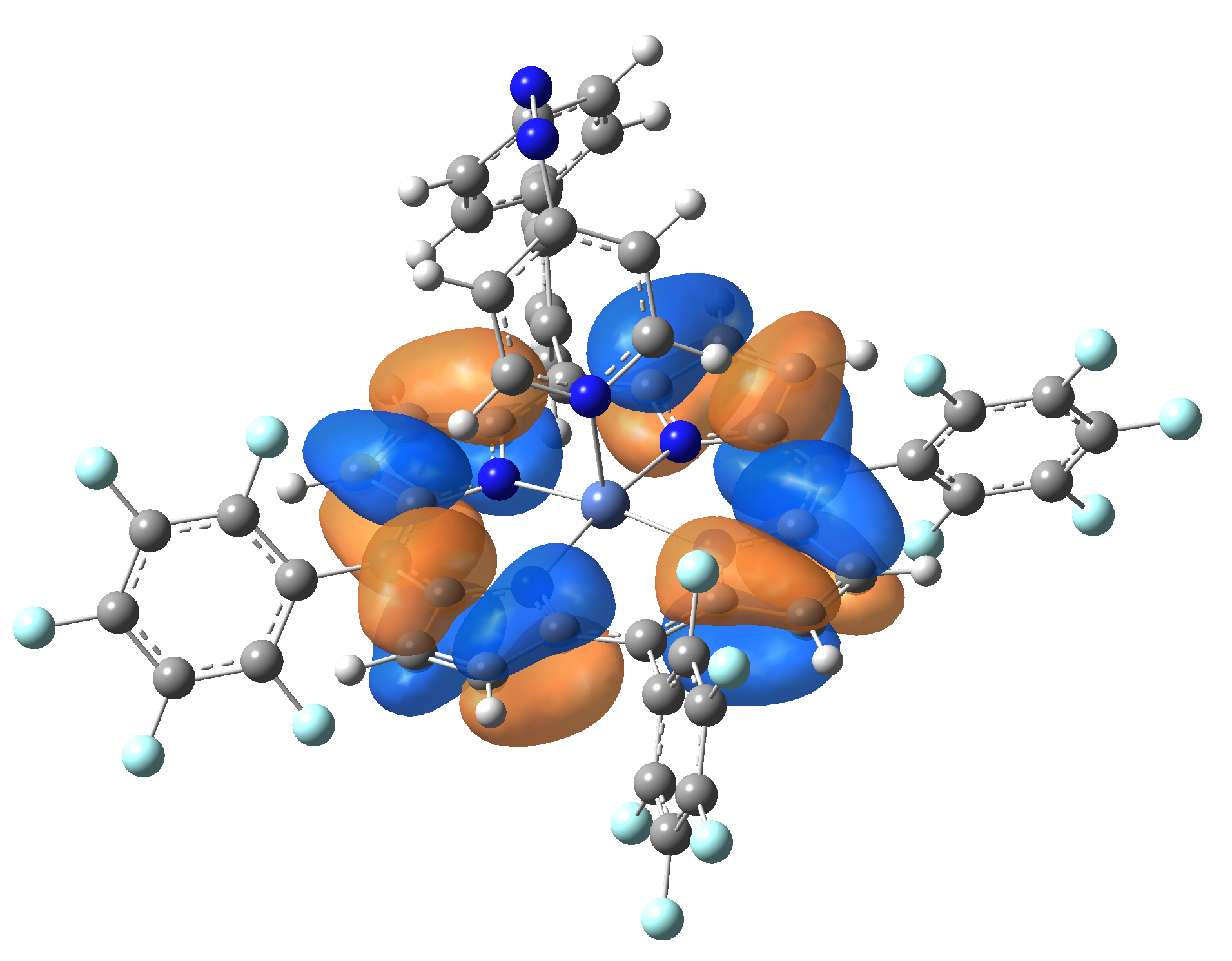 |
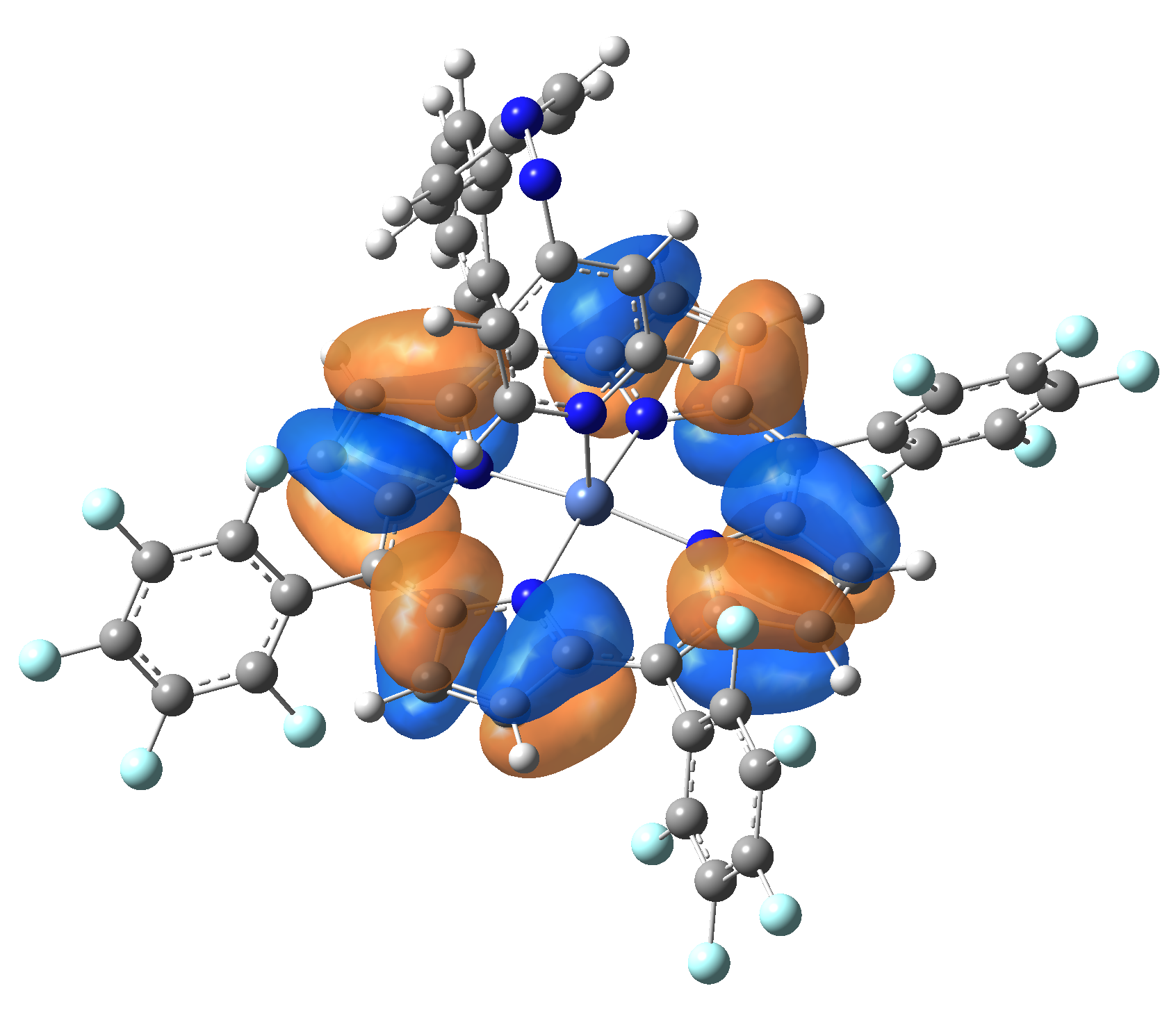 |
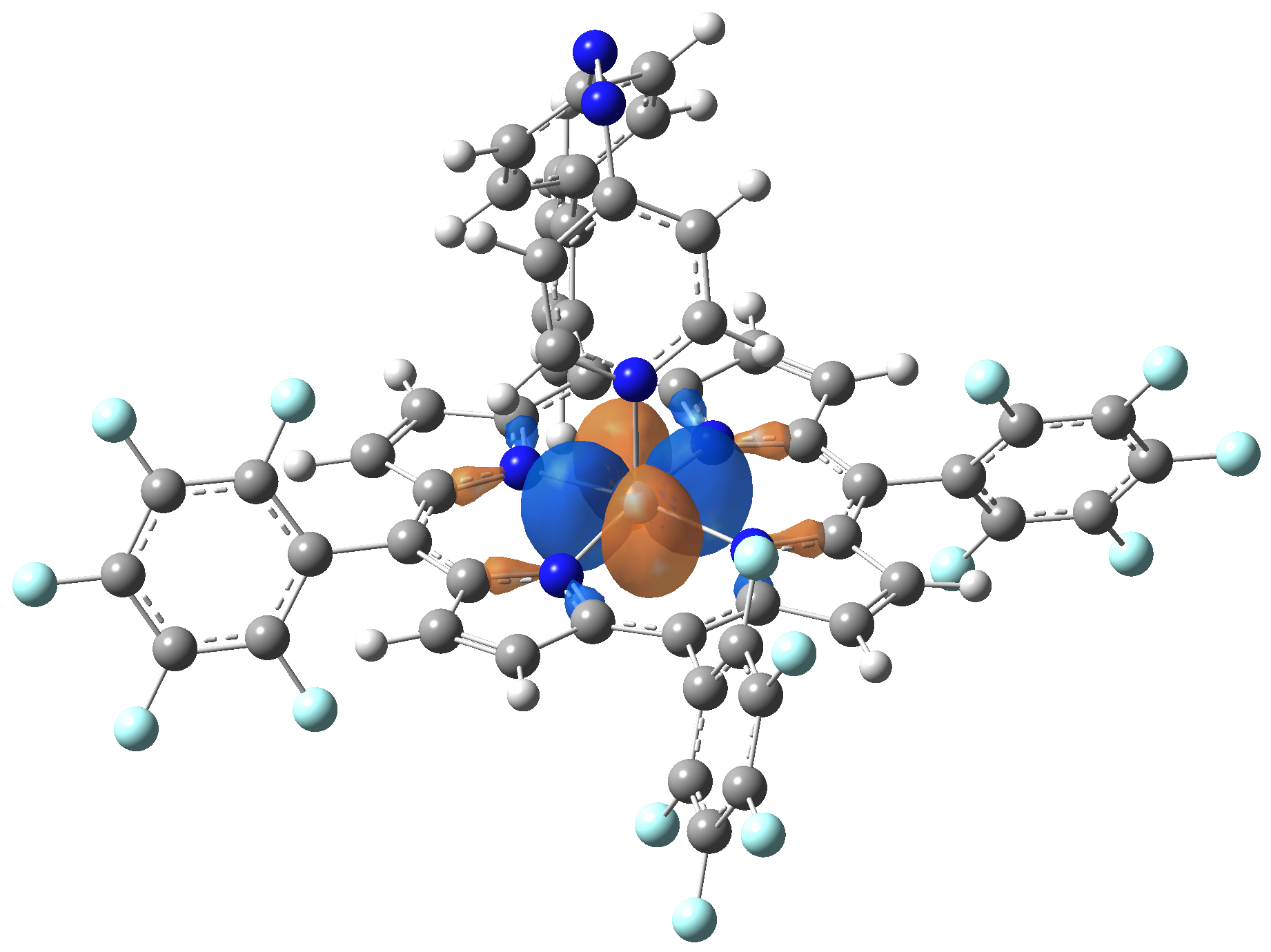 |
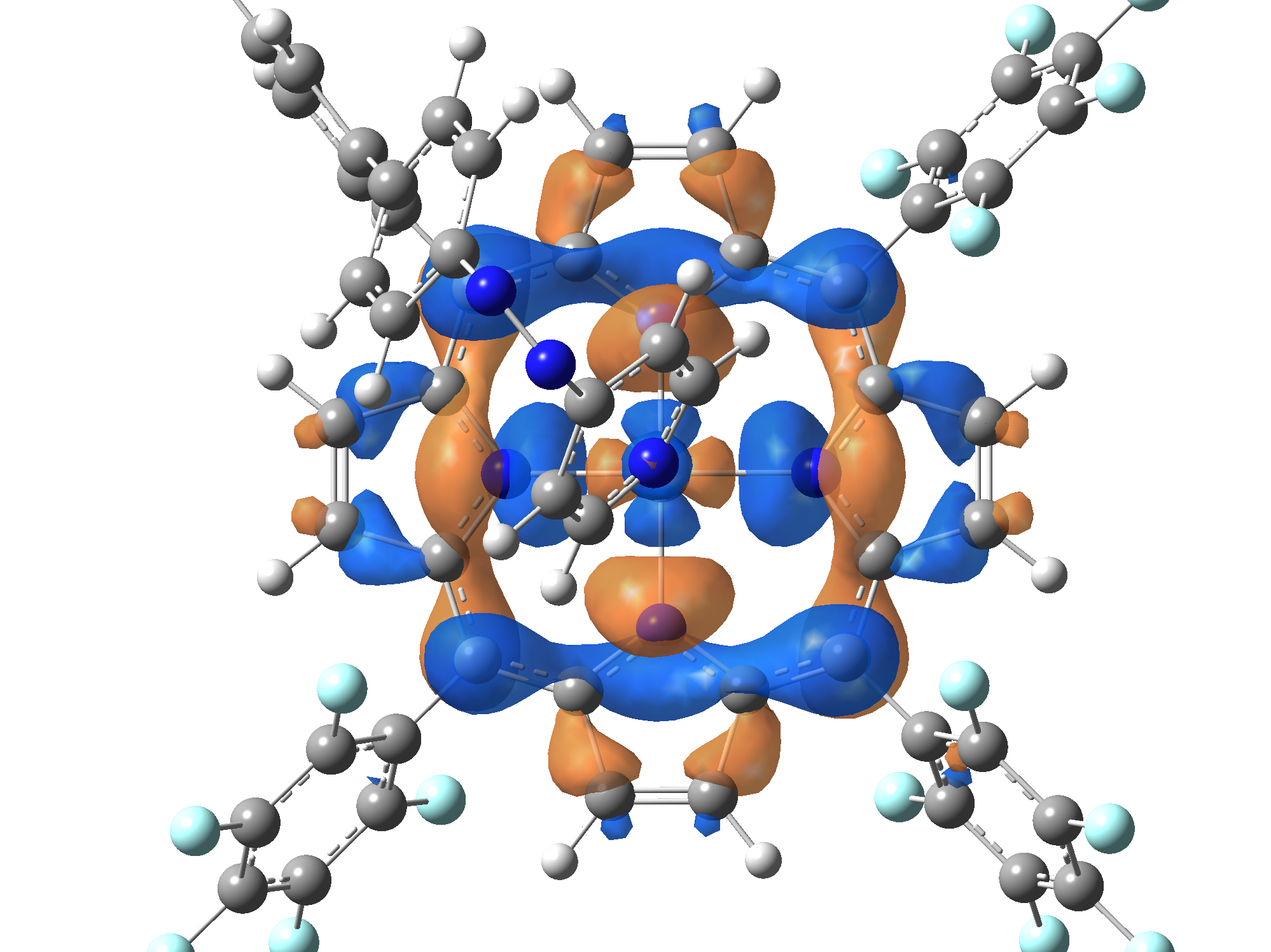 |
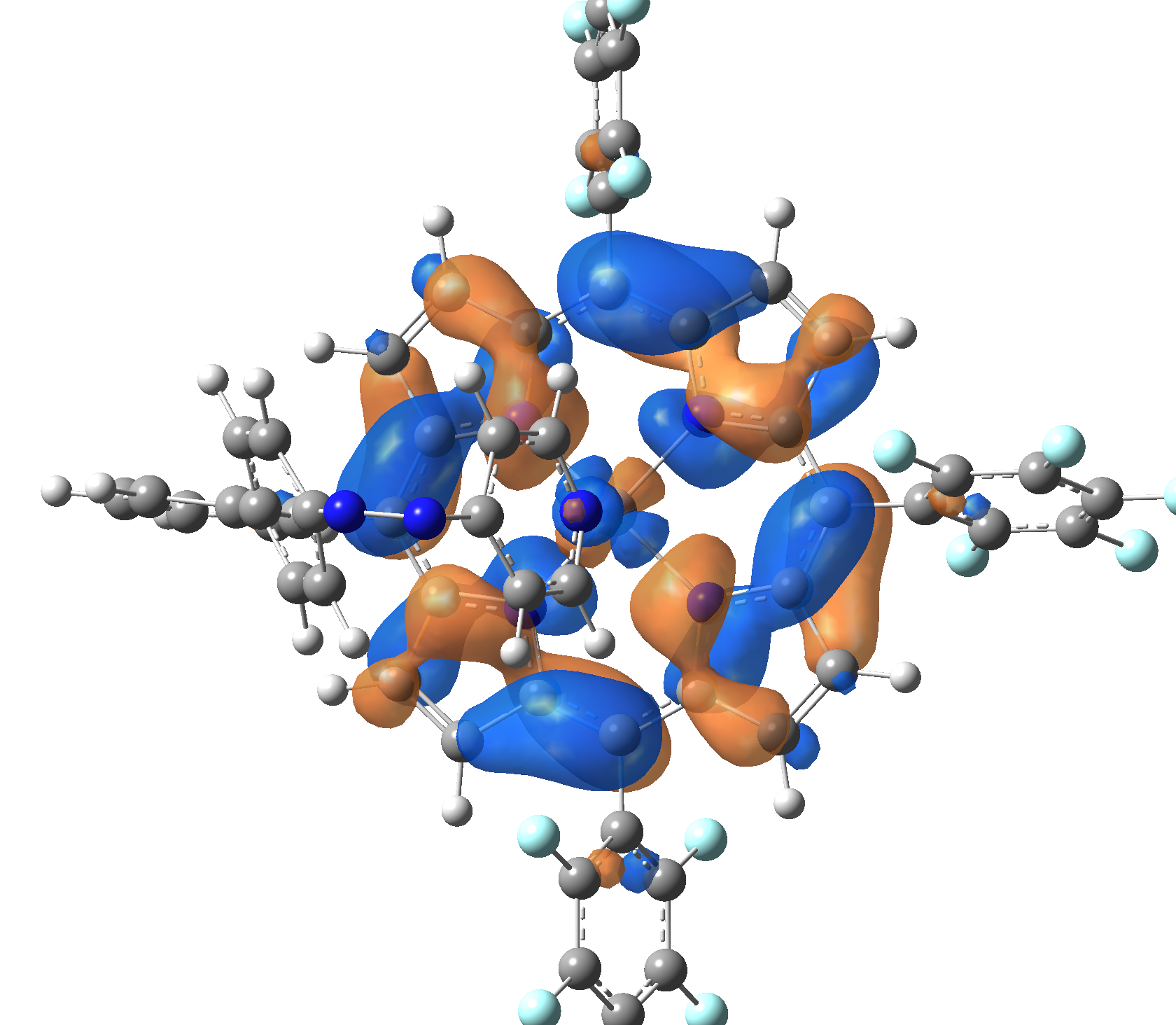 |
| LUMO | 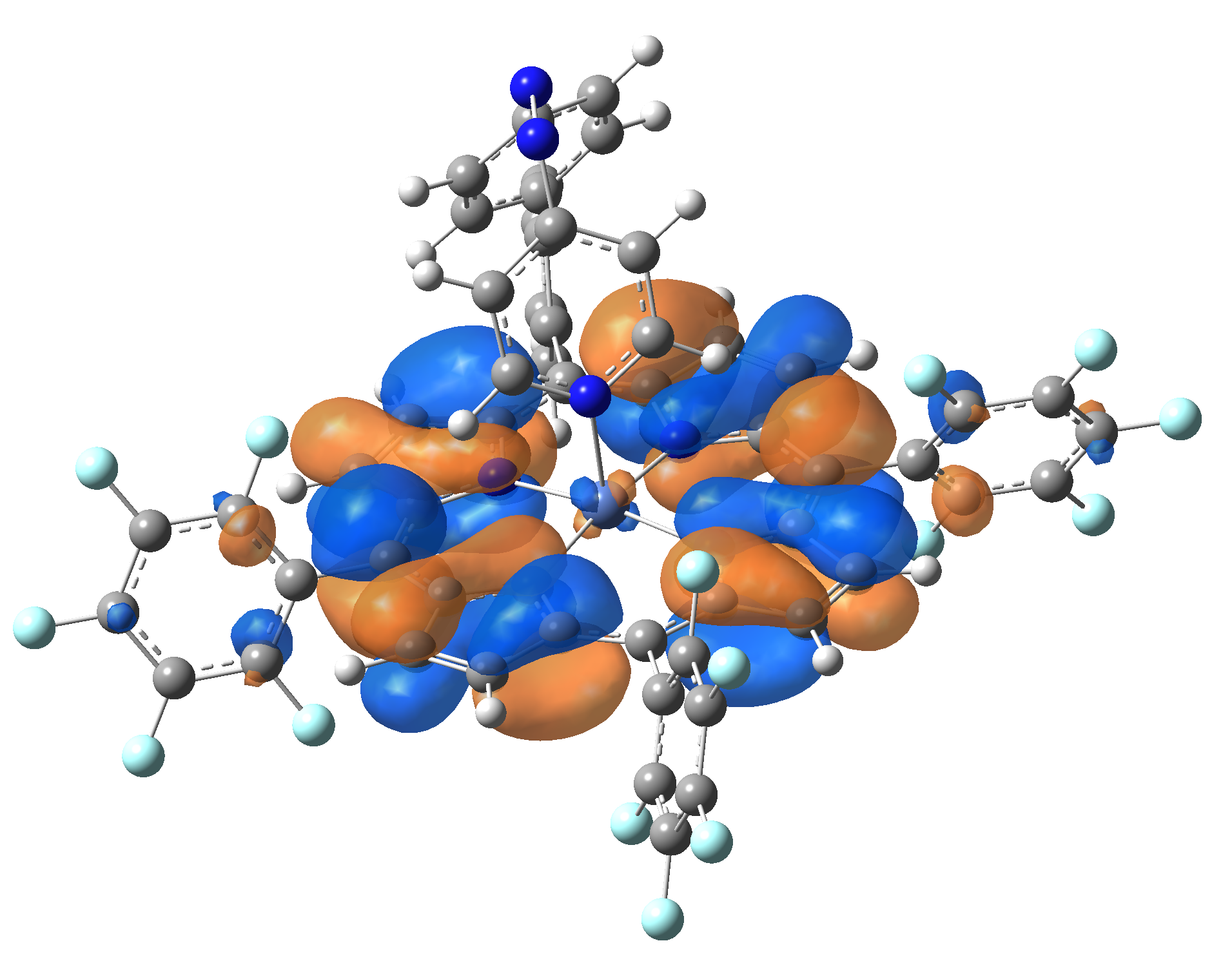 |
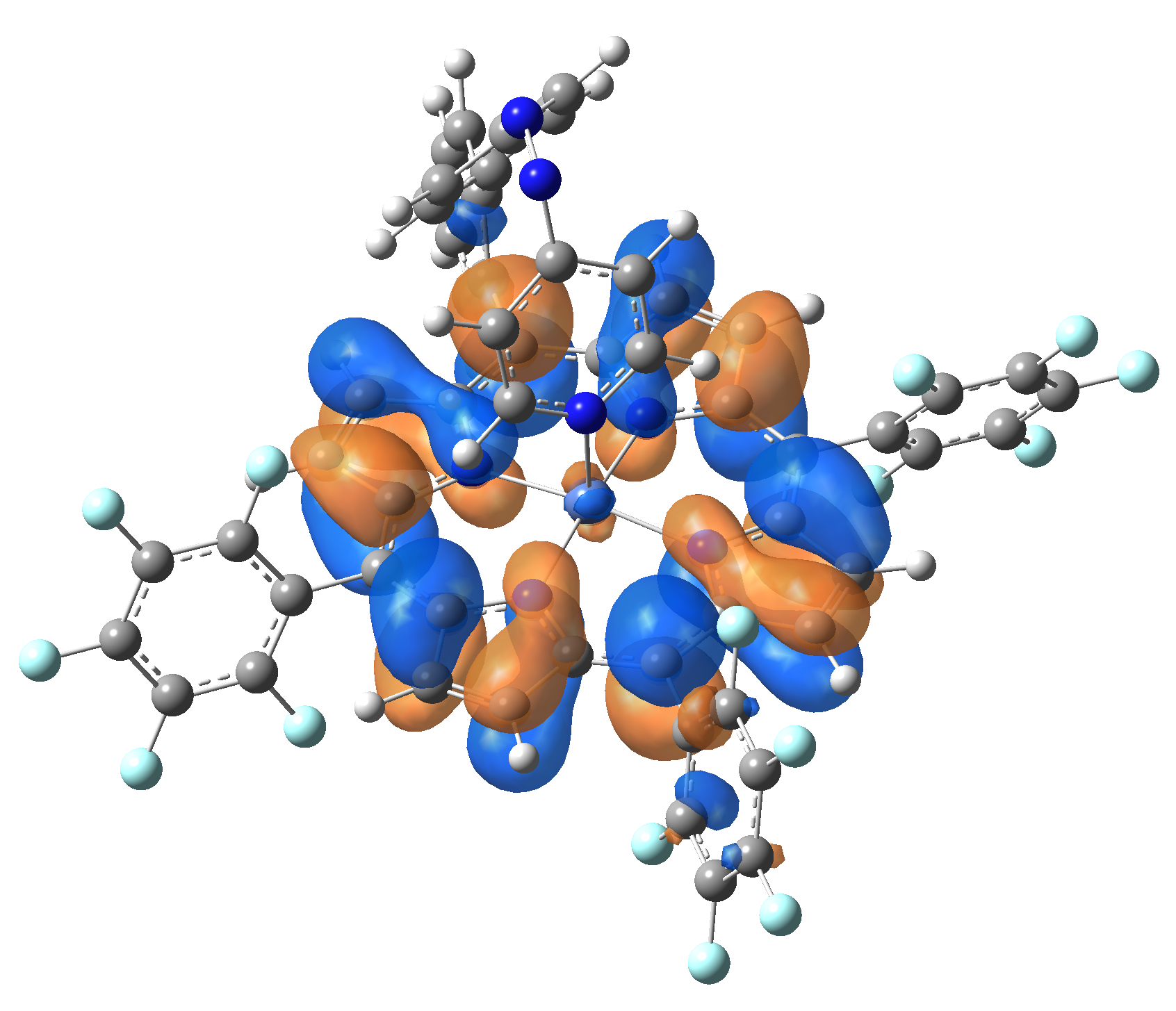 |
 |
 |
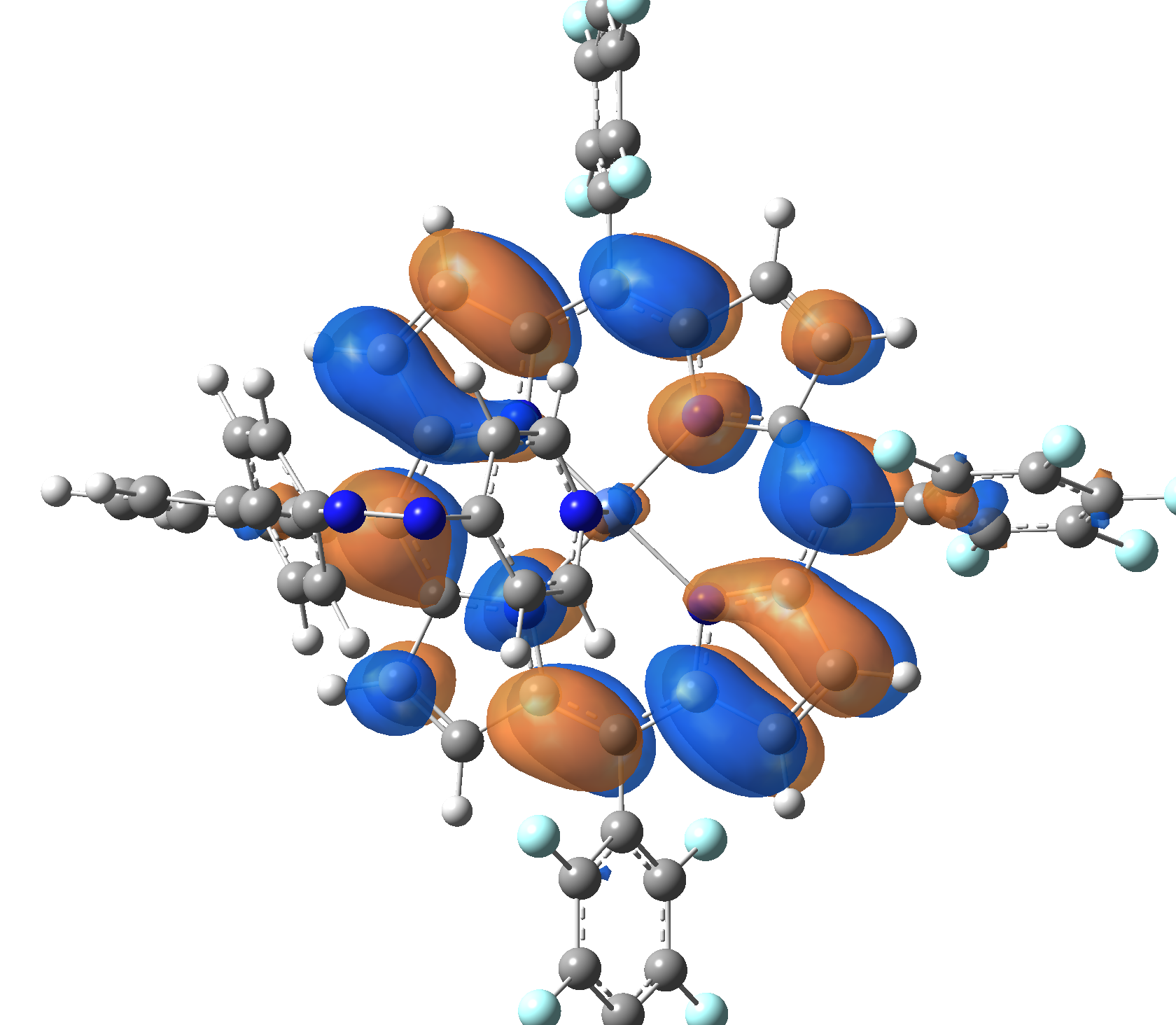 |
Table 2. The energy and oscillator strength as well as the HOMO and the LUMO NTO orbitals for the first 10 electronic transition with triplet spin state of the Ni-TPP-AP molecular complex.
As one can identifies from Tables 1 and 2, the first three transitions are the d ↔ d transitions which are forbidden due to the so-called "Laporte rule". The first active excited electronic state with singlet spin configuration is the S4 state, but its oscillator strength is still small in order to have a good absorption efficiency. Therefore the S5 electronic state is more suitable for the efficient excitation of our molecular complex. Similarly, the electronic excited state with most efficient laser field absorbtion is obtained for the T9 triplet spin state. In this way the laser induced singlet - triplet spin transition is compiled from the following parts: ① S0 → S6 with λ=475.82 nm; ② S6 ⊗ T4; ③ T4 ⟿ T0, while the backward transition is: ① T0 → T14 with λ=433.60 nm; ② T14 ⊗ S6; ③ S6 ⟿ S0.
Ni-P-biAP. Considering the same detailed analysis of the vertical electronic excitation levels we have concluded that the electronic excited states with most efficient laser field absorbtion are obtained for the S4 singlet and T10 triplet spin states, respectively. The laser induced fore and back transitions are: ① S0 → S4 with λ=490.59 nm; ② S4 ⊗ T10; ③ T10 ⟿ T0 and ① T0 → T10 with λ=525.06 nm; ② T10 ⊗ S4; ③ S4 ⟿ S0, respectively.
O2 - |
Searching for different ligand structures in order to get high efficient and well-controlled "Low-spin" and "High-spin" transitions; (Done) |
M04 - |
Computing the vertical excitation energies and the theoretical UV absorption spectra for different hypothetically assembled macrocycles with wellchosen molecular fragments, considering at least 5-6 possible candidates; (Done) |
M05 - |
Searching for the intersystem crossing point and computing the spin-orbit coupling in case of previously studied macrocycles as well as planning the macrocycle synthesis for the most promising spin crossover complexes; (Done) |
M06 - |
Drawing general conclusions about the spin crossover performance of different macrocycles with respect of the used molecular fragments; (Done) |
The equilibrium geometry structure analysis
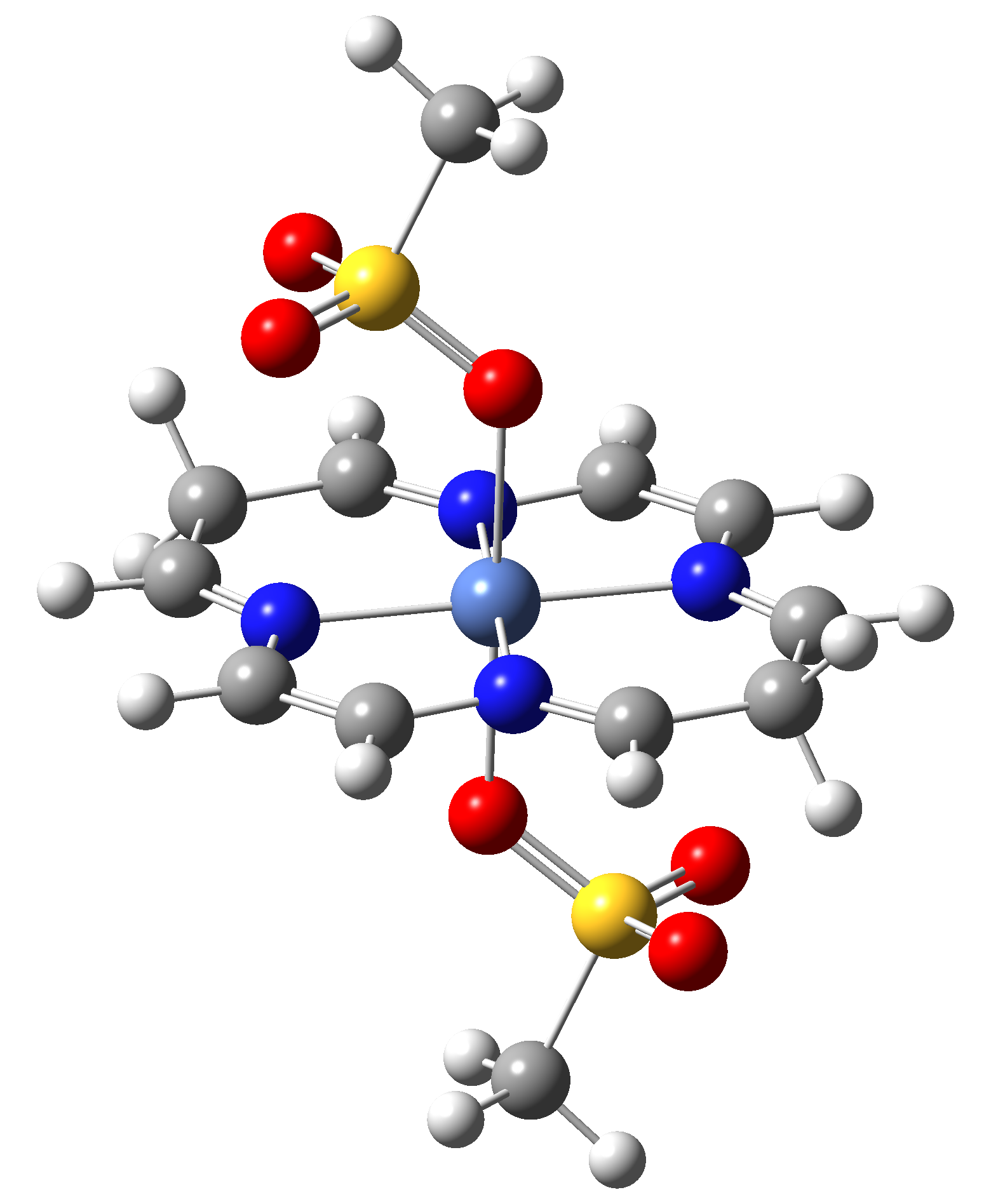
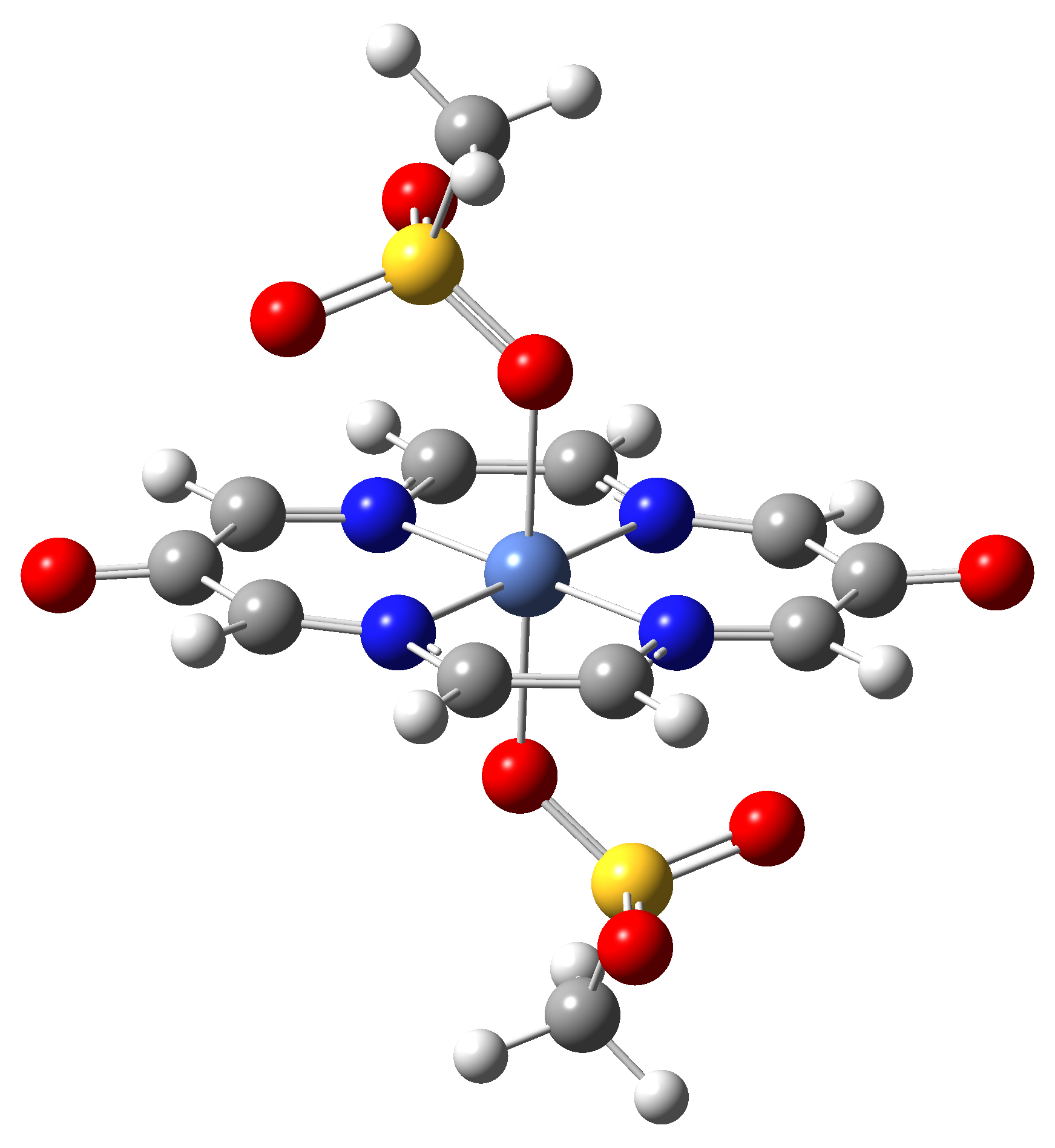
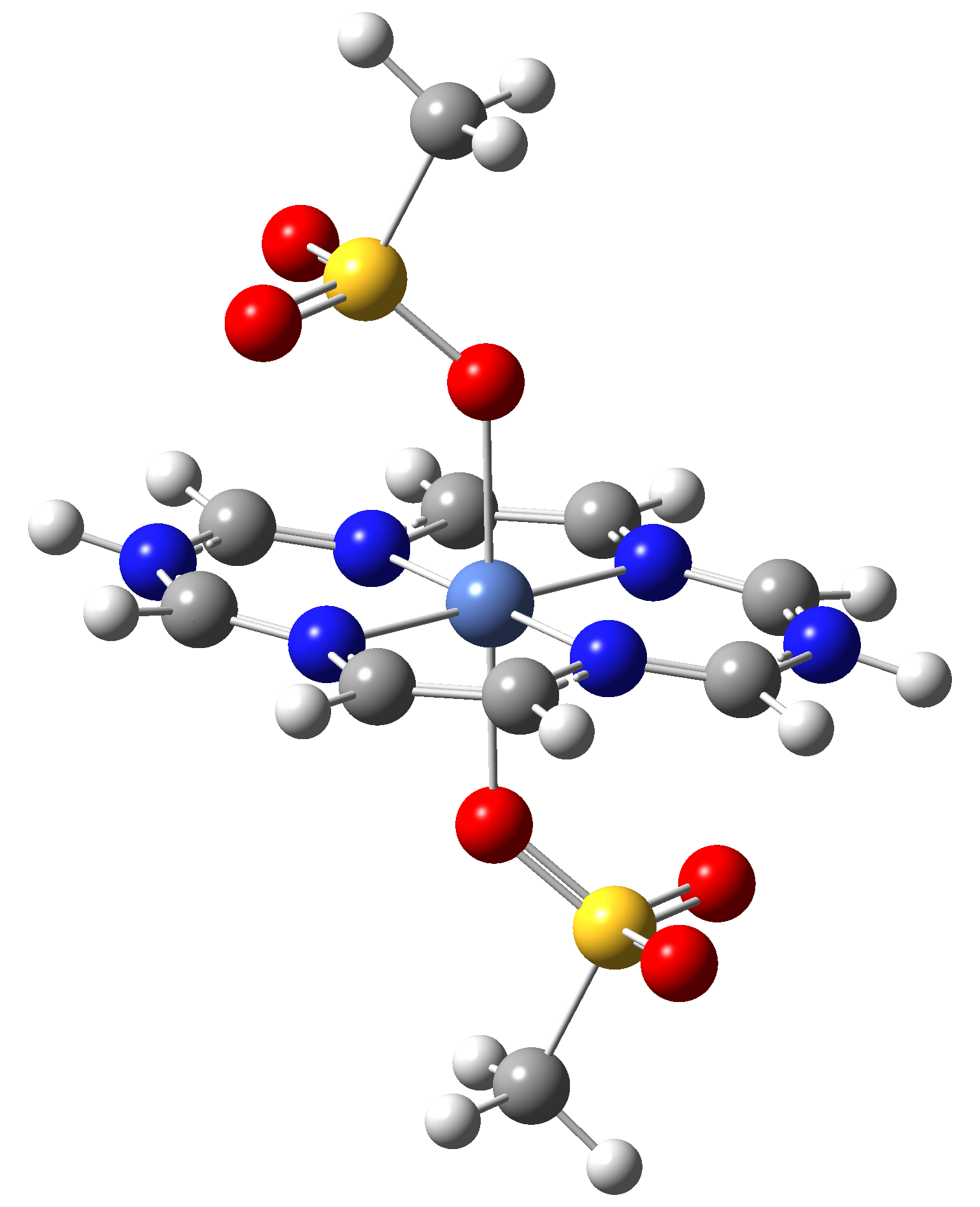
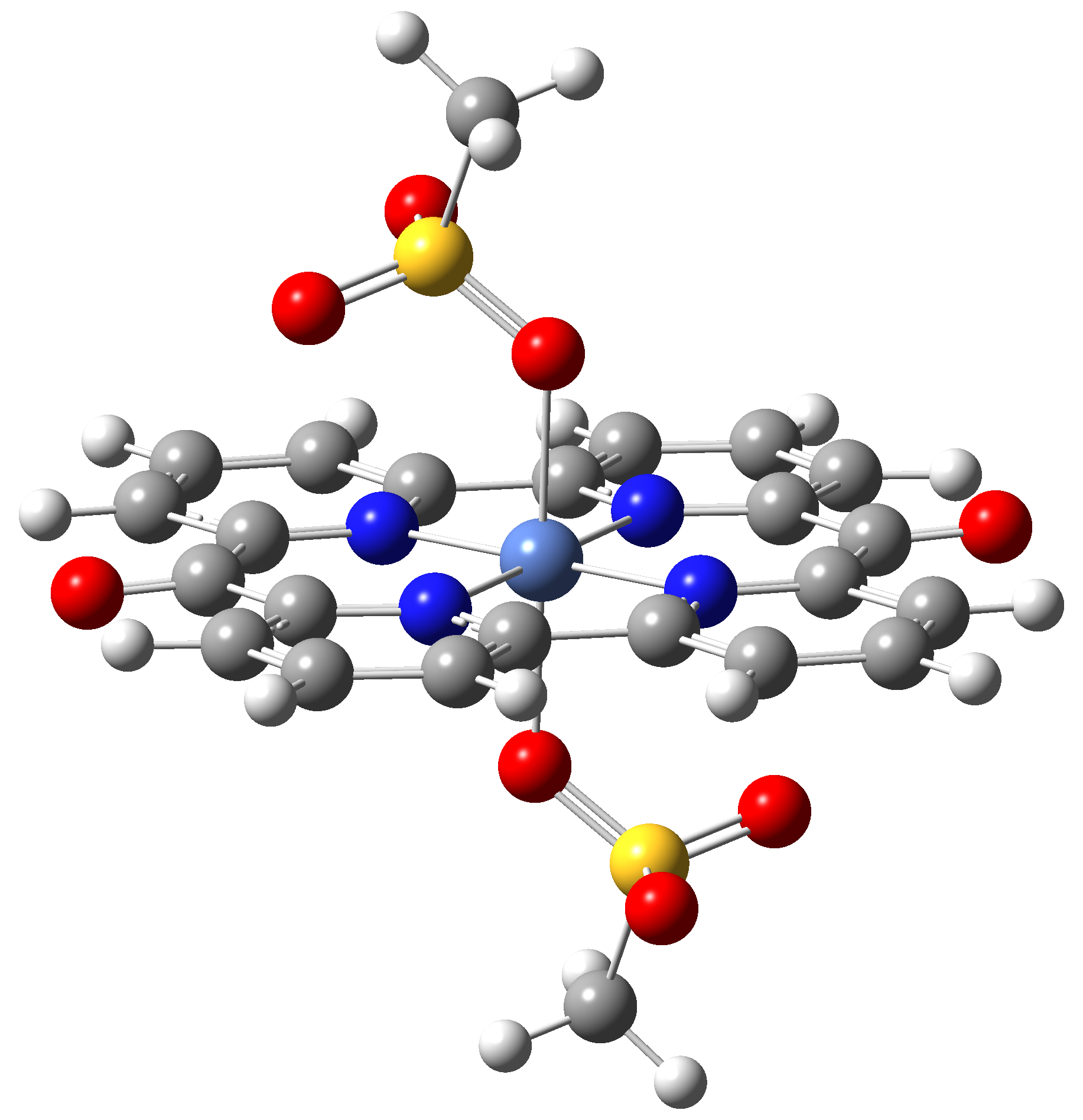
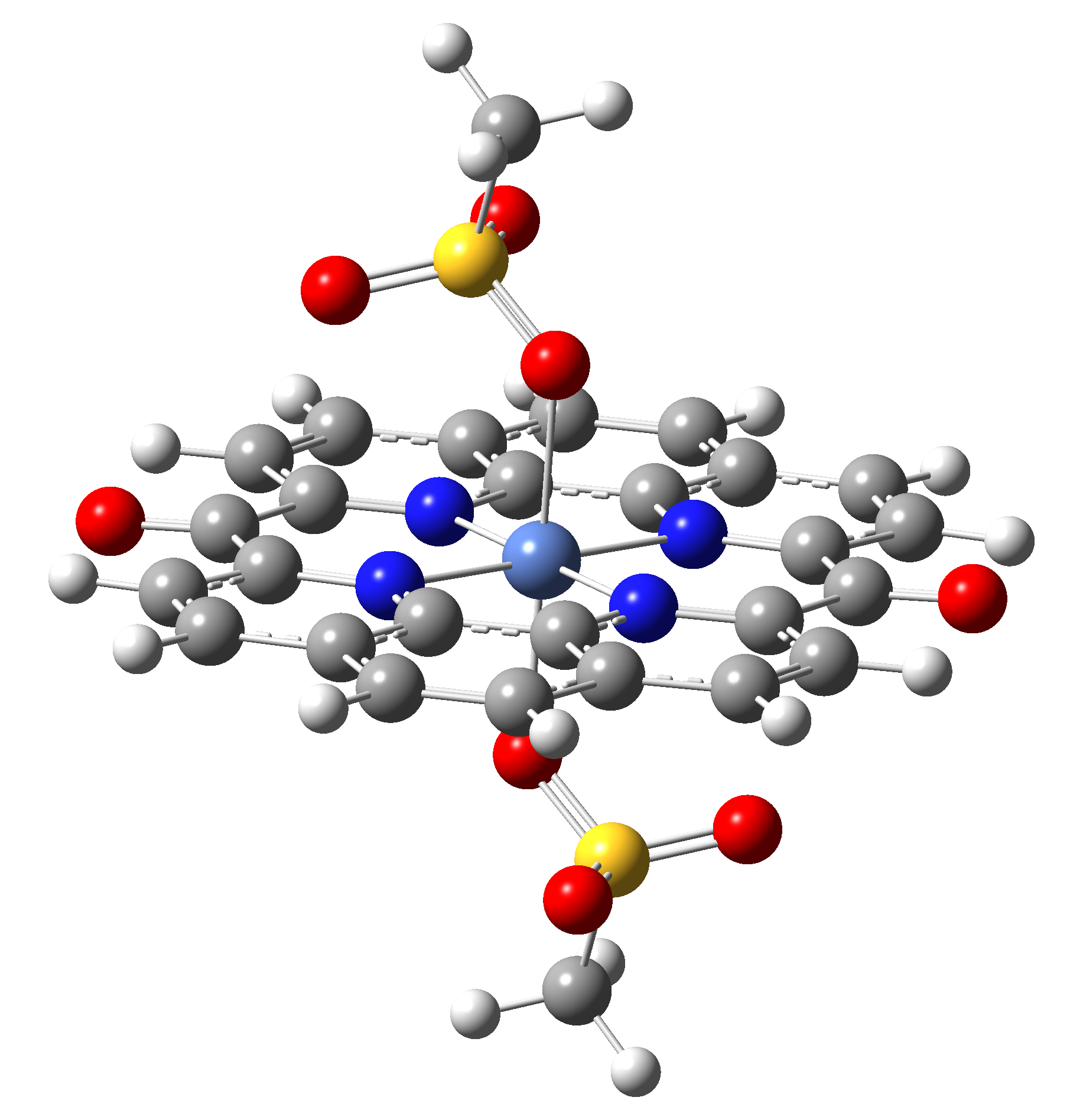
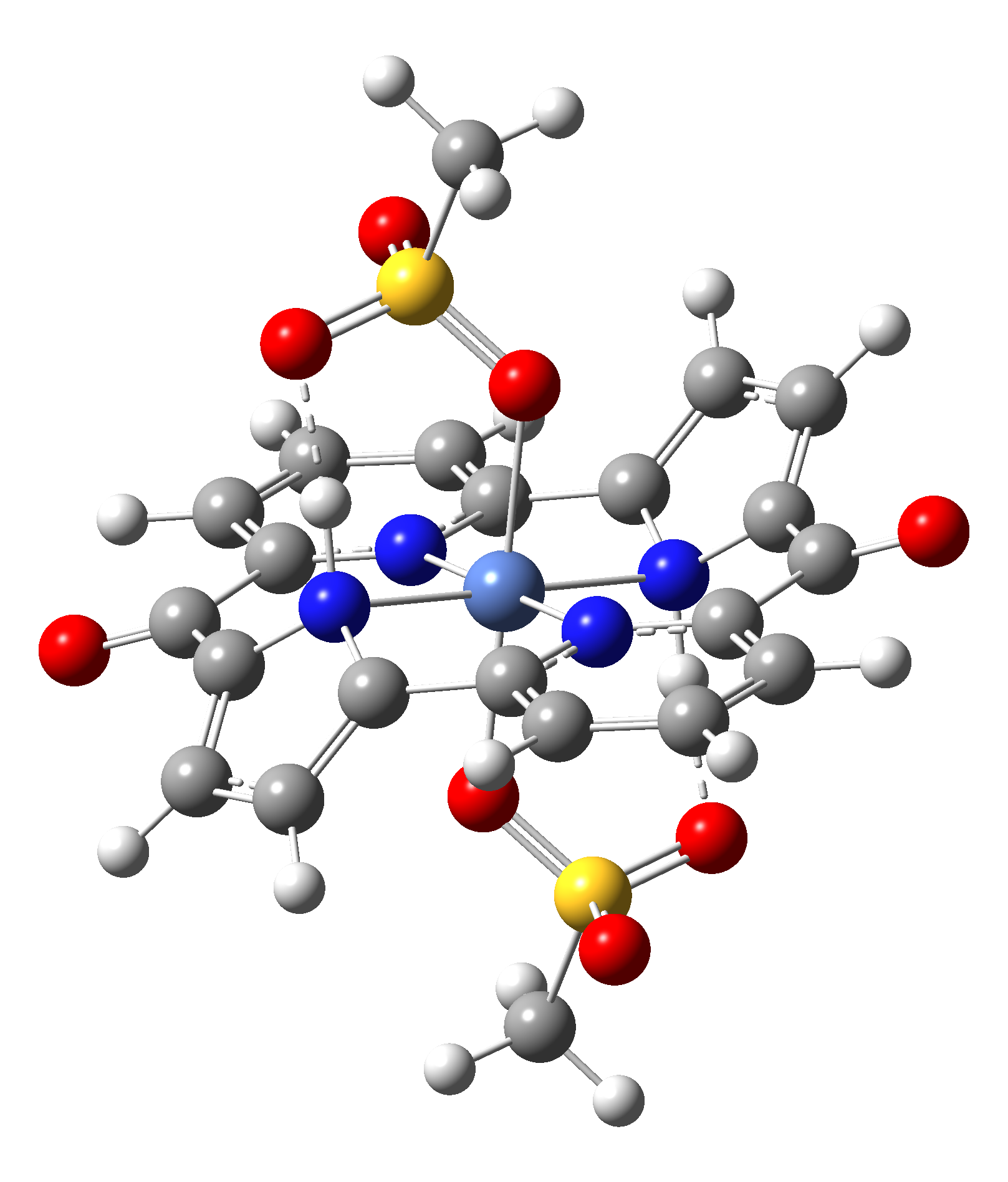
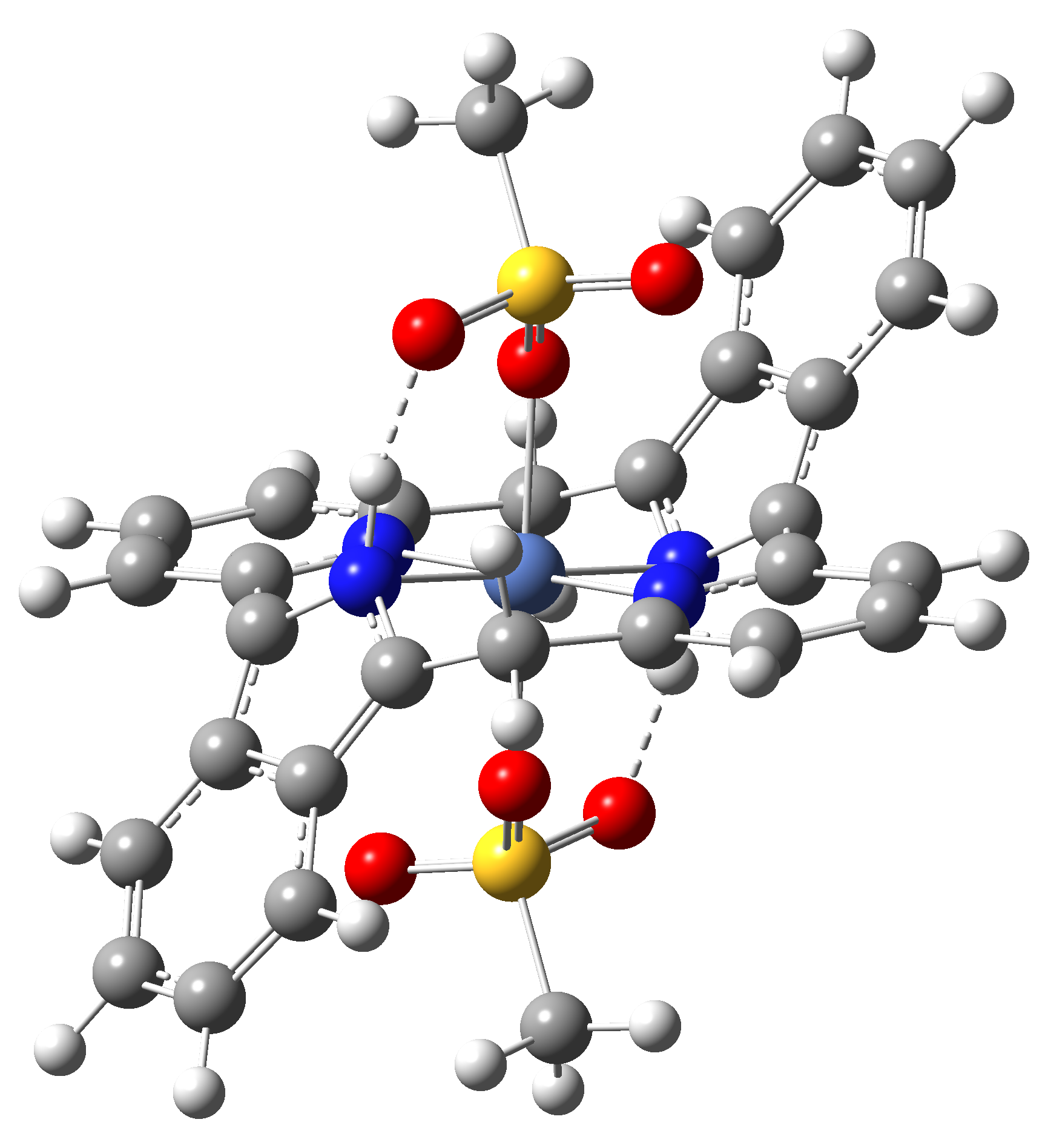
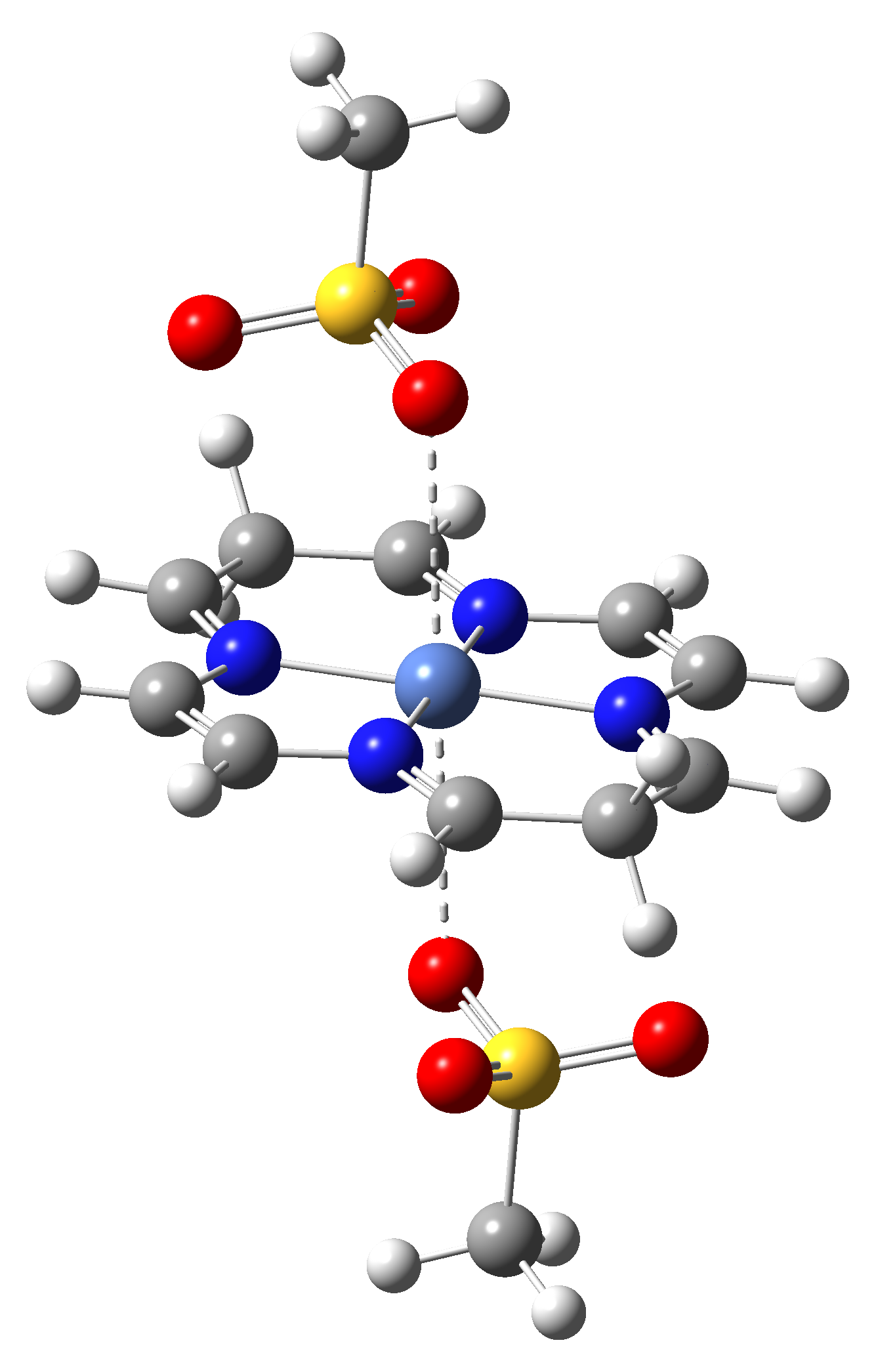
Based on the conclusions drawn from the analysis of the spin crossover performance of Ni-TPP-AP and Ni-P-biAP molecular complexes as well as from our previous experience presented in Ref. [2] we have tailored eight different supramolecular complexes for the studying the spin crossover efficiency (for example, accessible rang of UV domain, good absorption efficiency, strong spin-orbit coupling, large volume deformation). The proposed structures are shown in Table 3.
| Struct 1 | Struct 2 | Struct 3 | Struct 4 |
|---|---|---|---|
 |
 |
 |
 |
| Struct 5 | Struct 6 | Struct 7 | Struct 8 |
 |
 |
 |
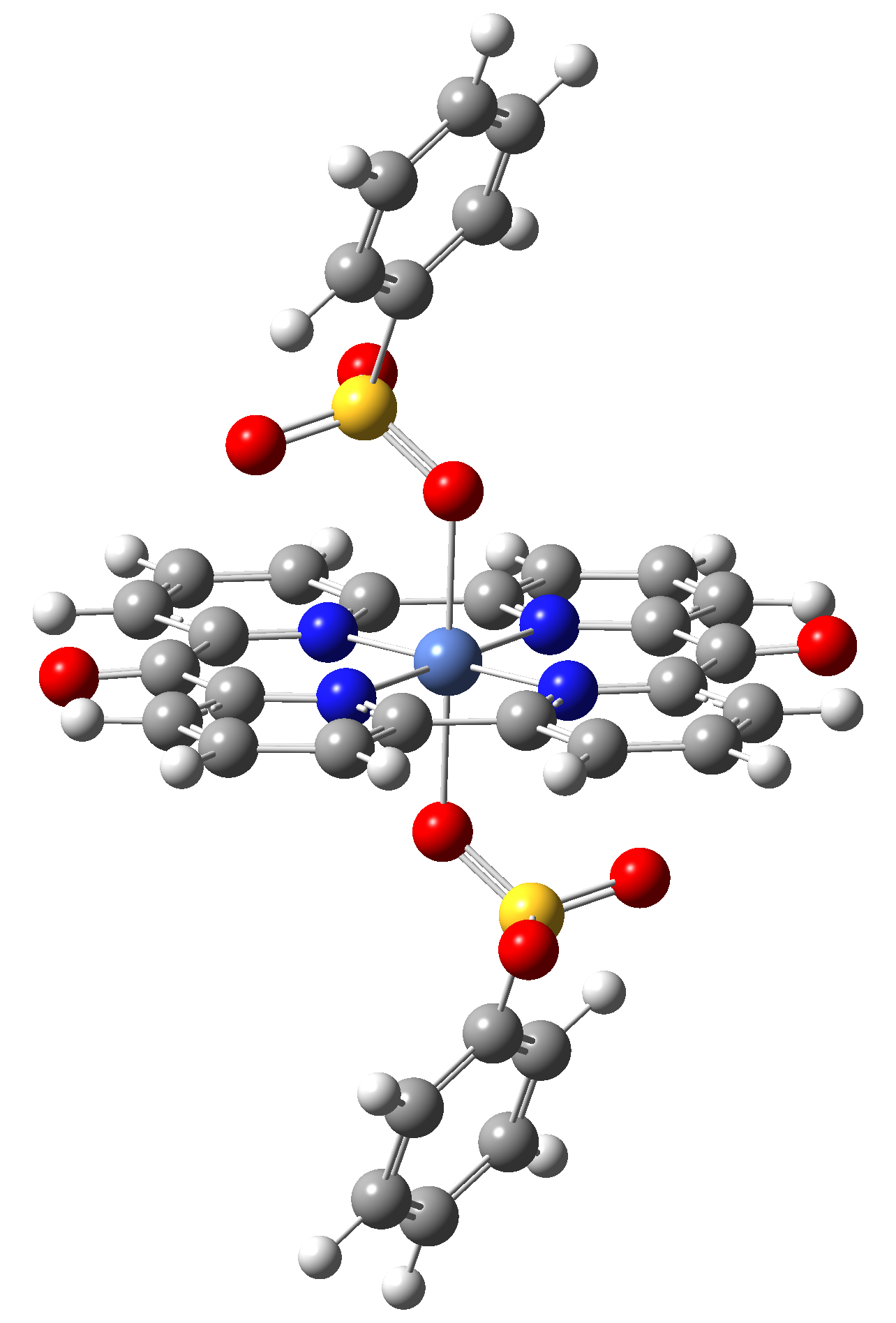 |
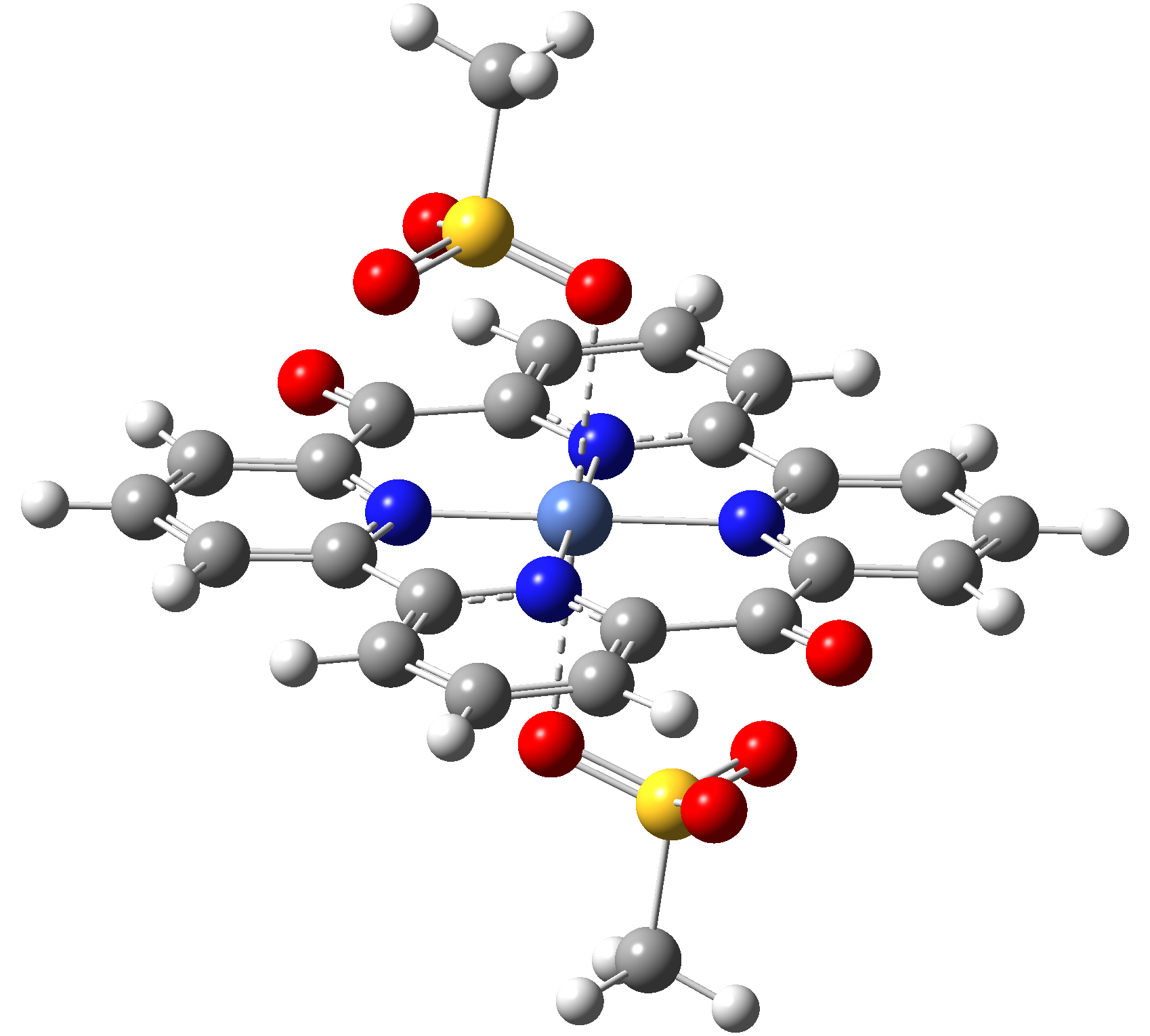
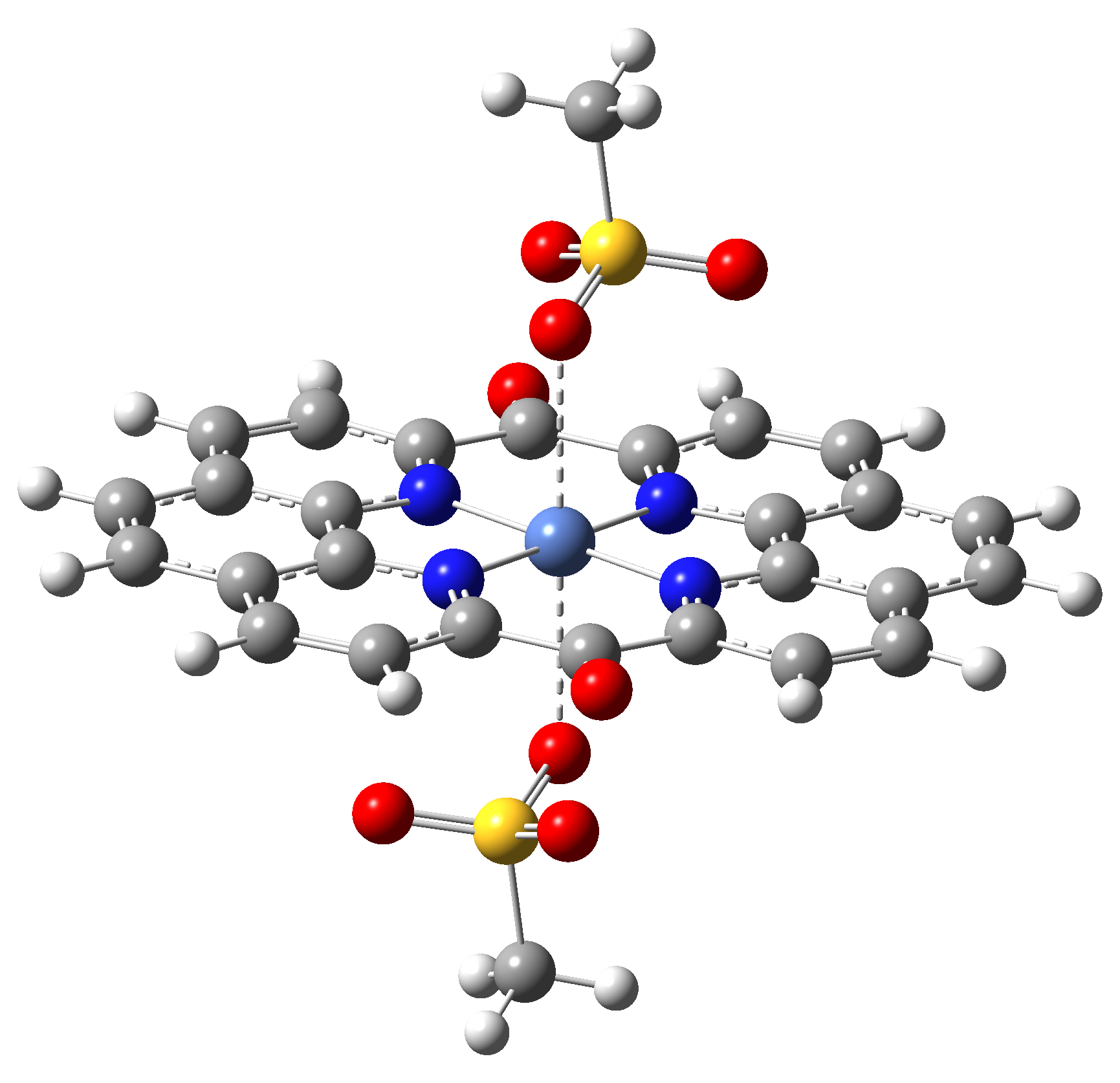
Table 3. The triplet state equilibrium geometries of eight different proposed molecular structures for studying the efficiency of the spin crossover phenomenon.
| Struct 1 | Struct 2 | Struct 3 | Struct 4 |
|---|---|---|---|
 |
 |
 |
 |
| Struct 5 | Struct 6 | Struct 7 | Struct 8 |
 |
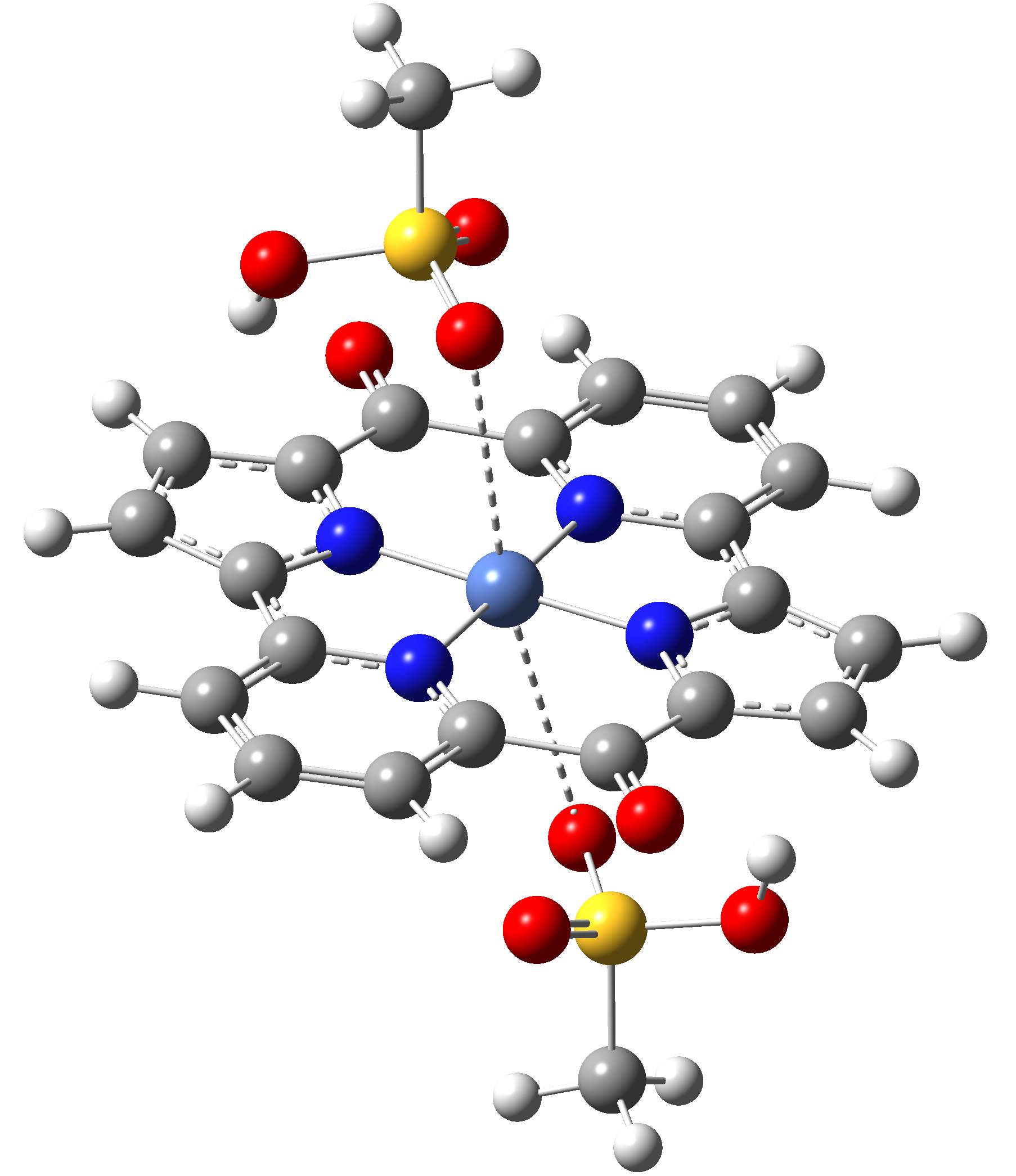 |
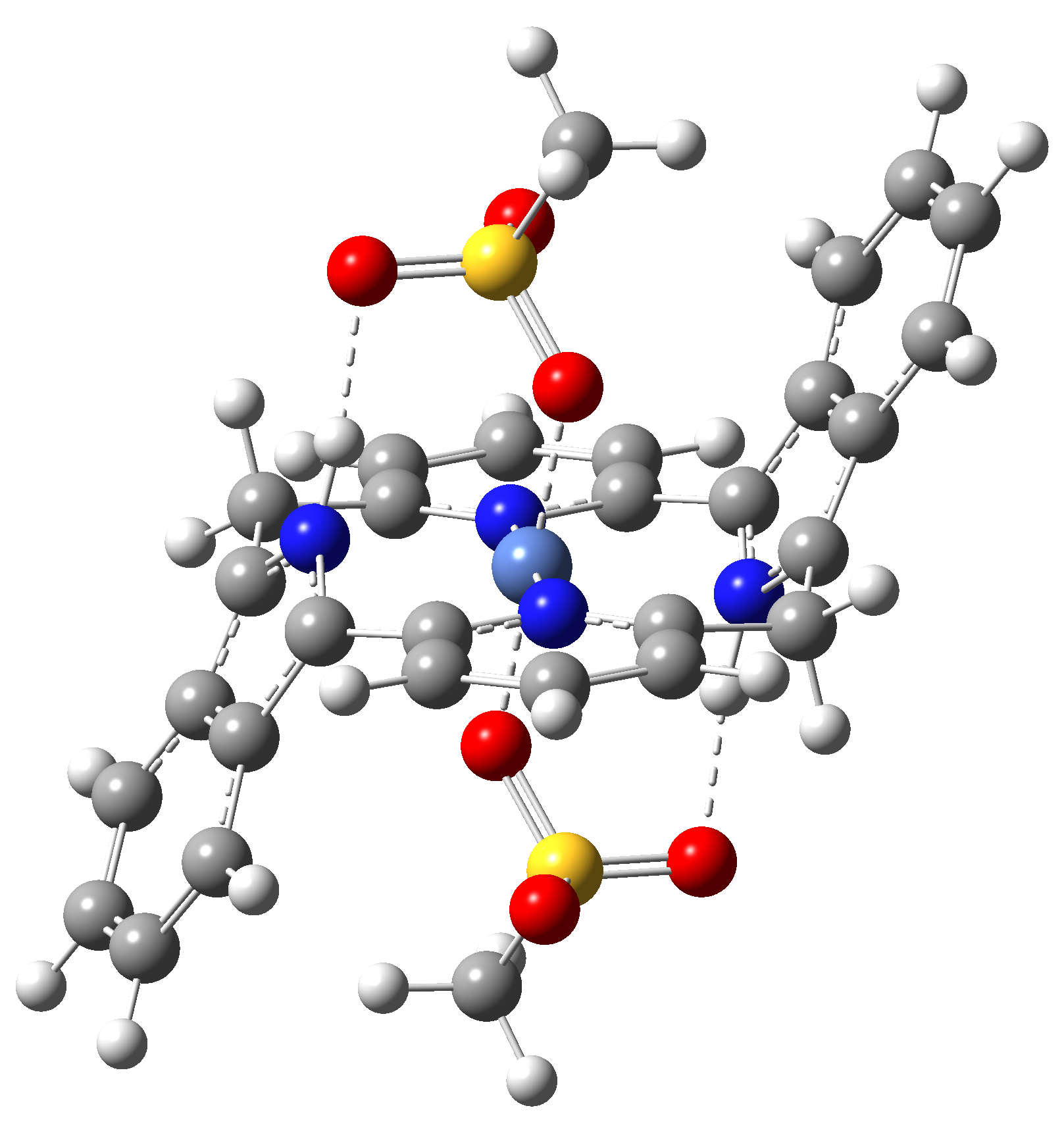 |
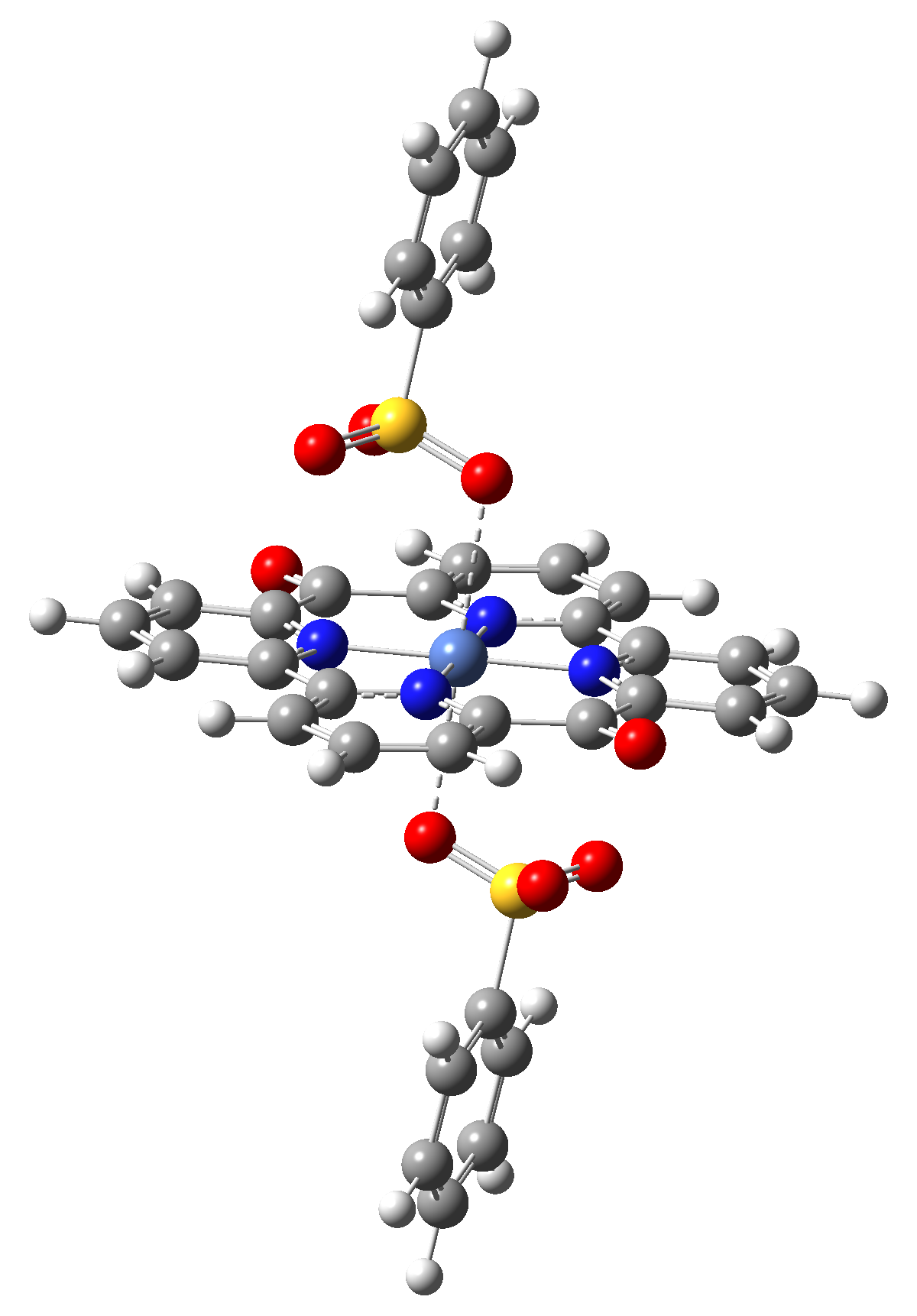 |
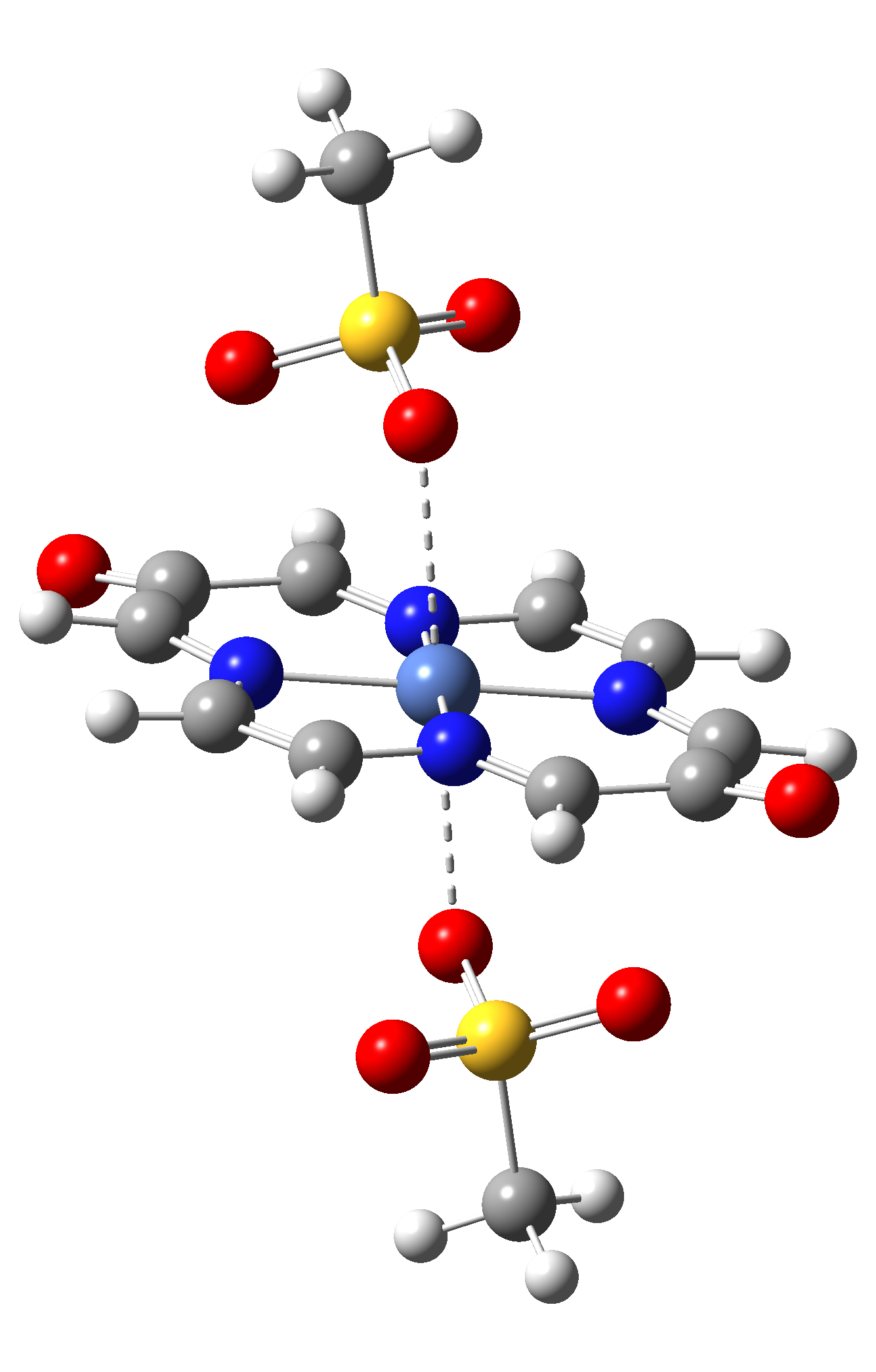
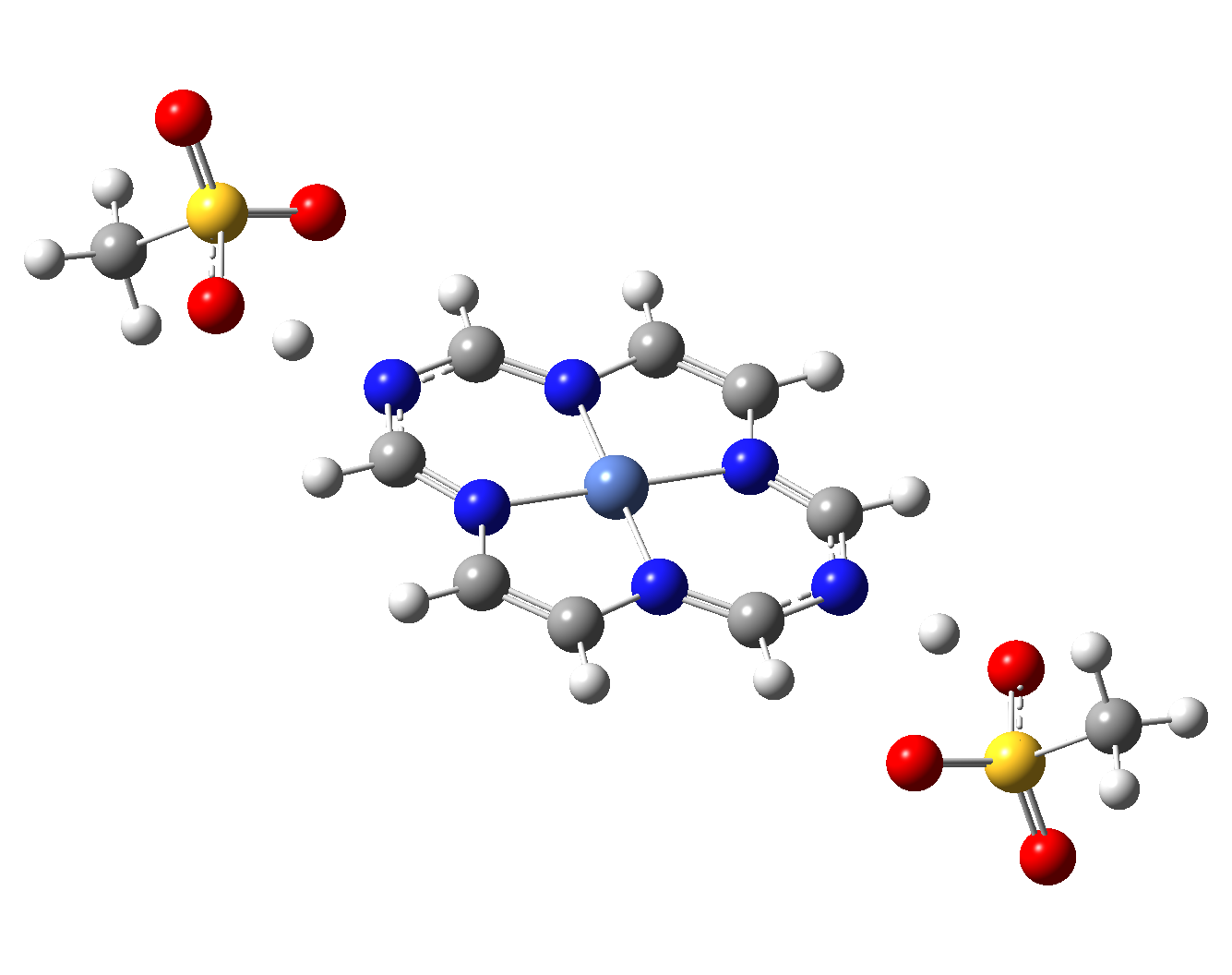
Table 4. The singlet state equilibrium geometries of eight different proposed molecular structures for studying the efficiency of the spin crossover phenomenon.
It can be observed that the singlet state equilibrium geometry with octahedral coordination configuration is destroyed in the case of Struct 3, while in the case of Struct 6 there is a proton transfer from the Nitrogen atoms of the ligand to the oxygen atoms of the mesylate (CH3-SO3-) anion fragments. Therefore, they will be excluded from our further investigation.
| Struct 1 | Struct 2 | Struct 4 | Struct 5 | Struct 7 | Struct 8 | |
|---|---|---|---|---|---|---|
| ESinglet (H) | -3442.34047057 | -3590.33428394 | -4050.14601171 | -4202.61336492 | -4133.11431782 | -4433.58368205 |
| RS(Ni···O) (Å) | 2.811 | 2.640 | 2.612 | 2.609 | 1.924 | 2.593 |
| ETriplet (H) | -3442.34801587 | -3590.35171473 | -4050.16632794 | -4202.62864037 | -4133.14474166 | -4433.60380049 |
| RT(Ni···O) (Å) | 2.136 | 2.109 | 2.125 | 2.133 | 2.109 | 2.125 |
| ES-T Gap (eV) | 0.205 | 0.474 | 0.553 | 0.416 | 0.828 | 0.547 |
| Active Excited | S12 | S4 | S3 | S3 | S5 | S2 |
| State | (311.99 nm) | (599.94 nm) | (521.73 nm) | (546.55 nm) | (484.20 nm) | (554.67 nm) |
| T21 | T7 | T12 | T12 | T8 | T10 | |
| (308.77 nm) | (537.14 nm) | (401.51 nm) | (434.87 nm) | (463.46 nm) | (424.54 nm) |
Table 5. The singlet and triplet total energies (in Hartree), the d(Ni···O) bond distances for the singlet and triplet equilibrium geometries (in Å), the singlet-triplet energy gap (in eV) and the first active excited state for both singlet and triplet vertical electronic excitations in the case of the six chosen molecular complexes.
Based on the structural analysis of the equilibrium geometries and the nature of vertical excitation energies for the six (Struct 1, Struct 2, Struct 4, Struct 5, Struct 7 and Struct 8) one can conclude that the most performant geometry structures which can exhibit singlet-triplet spin transition induced by the external electromagnetic radiation are the Struct 2, Struct 4 and Struct 8 molecular complexes.
As next step we will analysis the form of the energy barrier defined by the ISC points on the potential energy hyper-surface. The ISC point of structure based on Struct_4 configuration was optimized using the PF algorithm. The d(Ni···O) bond distance for this ISC geometry is 2.243 Å, while the energy barriers are: Δ(ISC-T0)=3.24 eV and Δ(ISC-S0)=2.86 eV
| Geometry | Spin | Ni···O‾ | Wiberg | Ni···N | Wiberg | EConf |
|---|---|---|---|---|---|---|
| State | (Å) | BO | (Å) | BO | (eV) | |
| Singlet | S | 2.667 | 0.085 | 1.904, 1.904 | 0.438, 0.439 | 2.856 |
| 2.669 | 0.086 | 1.909, 1.909 | 0.466, 0.466 | |||
| Triplet | T | 2.122 | 0.261 | 1.989, 1.989 | 0.299, 0.299 | 0.0 |
| 2.122 | 0.261 | 1.993, 1.993 | 0.301, 0.301 | |||
| ISC | S | 2.243 | 0.165 | 1.893, 1.893 | 0.494, 0.494 | 3.238 |
| 2.243 | 0.165 | 1.900, 1.900 | 0.497, 0.497 | |||
| T | 2.243 | 0.204 | 1.893, 1.893 | 0.340, 0.340 | ||
| 2.243 | 0.204 | 1.900, 1.900 | 0.344, 0.344 |
Table 6. The characteristic ligand bond distances (in Å) and Wibergs bond order indexes between the Ni central atom and the nitrogen and oxygen atoms for conformations with singlet and triplet spin configurations as well as for the ISC geometry of octahedral complex based on Struct_4 ligand. The conformational energy differences (in eV) between different geometry conformations are also given in the last column.
It can be observed that the geometry of the equilibrium configuration for singlet spin state mainly shows a square-planar coordination configuration, since the Wiberg bond order (BO) indexes along the vertical coordination bonds are extremely weak, and it can hardly be considered as bonds, perhaps just a weak intermolecular attractive interaction.
From the stability point of view it is very important that the vertical CH3-SO3‾ (mesylate) ligands in the singlet spin configuration geometry do not move away from the central Ni(II) ion in order to keep the structural stability and implicitly the reversibility of the singlet ↔ triplet spin transition. Accordingly, the intermolecular interaction energy between the planar Struct_4 ligand-metal complex and the two mesylate groups as well as the electrostatic potential map around the planar Struct_4 ligand-metal complex has been also calculated. The interaction energy is 157.95 kcal/mol per mesylate group due to the strong charge transfer effects. The amount of transferred charge from the negatively charged mesylate groups to the positively charged planar ligand-metal complex is 0.11e per mesylate group. The 3D graphical form of the potential energy surface is shown in Figure 5.
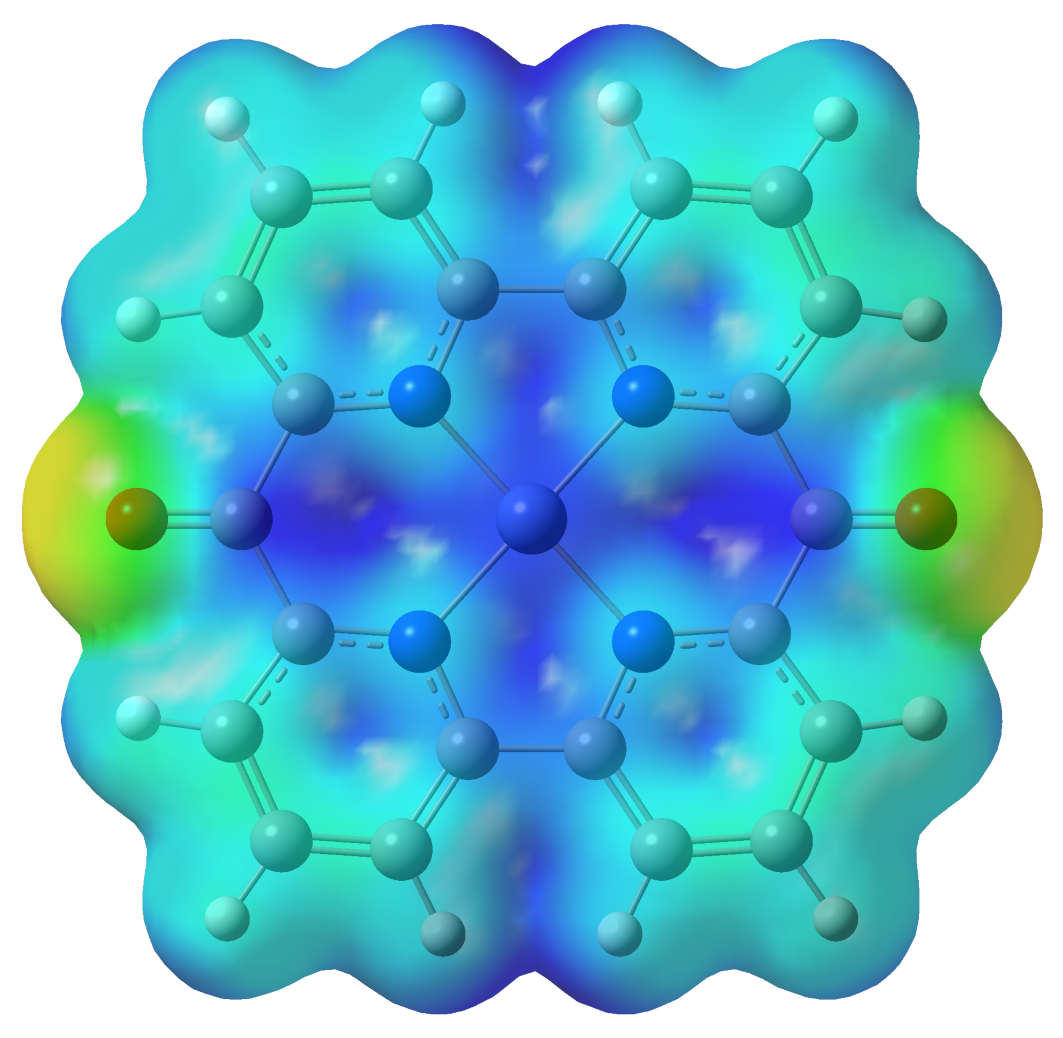
Figure 5. The map of the electrostatic potential (at 0.02 surface iso value) around the planar Struct_4 ligand-metal complex with Emin=-0.1e (red-yellow region) and Emax=+0.3e (dark blue region) charge values.
It can be observed from Figure 5, that the electron poor region (dark blue) is located around the central Ni(II) ion, which is an evidence of the fact that the planar Struct_4 ligand-metal complex will remain together with the two mesylate groups.
Based on the theoretical results regarding the structure stability of the Struct_4 ligand-metal complex, the energetic properties of its singlet ↔ triplet spin transition as well as the already existing synthesis model6, as first step, this octahedral coordination complex is proposed for chemical synthesis and further experimental investigations in the third period of the project implementation.
Conclusions
✔
For the Ni-TPP-AP and the Ni-P-biAP molecular complexes, using the MN12-SX/def2-TZVP level of theory, it has been drawn the energetical conditions for singlet ↔ triplet spin transition at ground state electronic level using the "Penalty Function" method and the Minimal Pathway constrained geometry optimization technique along the Ni···N bond. Both methods give the same Intersystem crossing (ISC) geometry.
✔
For the Ni-TPP-AP and the Ni-P-biAP molecular complexes, using the Breit-Pauli Hamiltonian, it was calculated the spin-orbit coupling taken in the geometry configuration of the ISC points of the potential hyper-surfaces. The relatively strong couplings show that the probability of the spin transition is significant;
✔
For the Ni-TPP-AP and the Ni-P-biAP molecular complexes, using the TD-DFT method, it was identified those "active" excited states which can efficiently absorb the laser radiation and which might be involved in the singlet ↔ triplet spin transition between different excited electronic states; The active states are: S6 singlet and T14 triplet electronic excited states;
✔
Based on the conclusions drawn for the Ni-TPP-AP and the Ni-P-biAP cases of studies, eight new organometallic structures were designed for further investigations as possible new spin crossover molecular complexes;
✔
Among the eight proposed organometallic complexes, for four structures it was demonstrated that they show adequate physical properties for the spin crossover phenomena. This was obtained i) by structural stability studies of the singlet and triplet equilibrium geometries, ii) by the localization of the ISC points on the potential hyper-surfaces and iii) by the identification of the "active" electronic excited states which can efficiently absorb the laser radiation;
✔
Struct_4 ligand-metal complex was proposed for chemical synthesis and further experimental investigations in the third period of the project implementation, but the partial results suggest that the Struct_5 structure might also presents spin crossover phenomena;
Bibliography:
C. Ciminelli, G. Granucci and M. Persico, Chem.-Eur. J. 10, 2327 (2004).
M. Vlassa and A. Bende, Chemical Physics, 457, 152_159 (2015).
S. Chiodo and N. Russo, J. Comput. Chem., 29, 912 (2008).
S. Chiodo and N. Russo, J. Comput. Chem., 30, 832 (2009).
S. G. Chiodo and M. Leopoldini, Comput. Phys. Commun., 185, 676 (2014).
E. Joliat-Wick, N. Weder, D. Klose, C. Bachmann, B. Spingler, B. Probst, and R. Alberto, Inorg. Chem., 57, 1651 (2018).